Document Type : Review Article
Authors
Department of Pharmaceutical Chemistry, St. Joseph’s College of Pharmacy, Cherthala, Kerala, India
Abstract
Imidazoles are key components of functional molecules with diverse applications. Due to its adaptive qualities in chemistry and pharmacology, its derivatives have attracted great study in recent years. The rapid growth of imidazole-based medicinal chemistry indicates the promising and potential therapeutic values of imidazole-derived compounds for treating irremediable diseases. This review is aimed on advances in the pharmacological activities of imidazole during past years such as anticancer, antioxidant, antibacterial, antifungal, antiviral, anti-tubercular, and anti-parasitic activity.
Graphical Abstract
)
Keywords
Main Subjects
- Introduction
Imidazole (1) is a five-membered aromatic molecule that contains two annular nitrogen atoms [1]. It has the chemical formula C3N2H4 and is an organic molecule (Figure 1). It is a soluble white or colorless substance that turns water into a slightly alkaline solution. It is an aromatic heterocycle, categorized as a diazole in chemistry, and has meta-substituted nitrogen atoms that are not next to each other. In recent years, interest in imidazole and its derivatives has increased due to their useful features in chemistry and pharmacology. The imidazole derivatives possess a wide range of biological activities such as antibacterial [2, 3], anticancer [4, 5], antitubercular [6, 7], antifungal [8], analgesic [9], antidiabetic [10], and antiviral [11].

Figure 1.
Two nitrogen atoms make up the imidazole nucleus, one of which has a hydrogen atom and the other is of the pyrrole type. A bio-ester of the pyrazole ring is the imidazole ring [12, 13].
Imidazole exhibits both acidic and basic properties due to its amphoteric nature. It is a solid that is either white or colourless and is very soluble in water as well as other polar solvents [14]. The imidazole ring is susceptible to attacks from both nucleophiles and electrophiles because of its amphoteric nature. The proton on the imidazole ring's first nitrogen atom is acidic [15]. It is an aromatic compound because the ring contains a sextet of electrons. Since glyoxal and ammonia were used in the original synthesis of imidazole, the compound was first known as glyoxaline [16].
Imidazole acts as an important pharmacophore in drug discovery (Figure 2). There are numerous instances of commercially marketed medications that include the imidazole ring on the market such as etomidate (4) (anaesthetic) [17], phenytoin (3) (anticonvulsant) [18], miconazole (antifungal) [19], metronidazole (2) (antiprotozoal) [20], azathioprine (7) (anti rheumatoid arthritis) [21], dacarbazine (5) (Hodgkin’s disease) [22], tinidazole (6) (antiprotozoal [23], and antibacterial [24], pimobendan (calcium sensitizer and phosphodiesterase inhibitor), etc.

Figure 2.
Medicinal properties of compounds containing imidazole include anticancer [25], analgesic, antifungal, antihypertensive [26], antiviral, antitumor, anti-tubercular, antihistaminic [27], anti-inflammatory [28], antidepressant [29], antidiabetic, anticonvulsant, anti-allergic, anti-rheumatic, etc. Herein, we provide a brief overview of the research status of imidazole-based compounds during the last 12 years with various biological activities including anti-cancer, antioxidant [30], antibacterial, antiviral, anti-tubercular, and anti-inflammatory.
The range of conditions that can be treated with imidazole medications in clinical medicine has increased. The subject of medicinal chemistry has a wide range of opportunities thanks to several ways imidazole can be synthesized. This article focuses on the research that has been done and the biological activity of imidazole over the last 12 years.
- Biological Activities
2.1. Anticancer activity
Cancer is a disease that manifests as the growth of aberrant cells with the capacity to penetrate and devastate healthy bodily tissue [31]. It is the leading global cause of death [32]. Imidazole and its derivatives have been utilized as anticancer drugs among other pharmaceutical uses. Imidazole has been claimed to have anticancer medication potential in numerous research studies. Following are a few of them.
IPM714 (8), a novel 1H-imidazole[4,5-f][1,10] phenanthroline derivative (Figure 3), was created, and it was discovered that it has inhibitory effects on colorectal cancer (CRC) cells specifically, with IC50 values of 1.74 M and 2 M in HCT116 cells and SW480 cells, respectively. The study demonstrated the IPM714cytotoxicity in numerous types of cancer cells as well as its in vitro anticancer mechanism. According to the cellular functional study, IPM714 can trigger apoptosis in HCT116 and SW480 cells and stop the HCT116 cell cycle in the S phase. Western blot and molecular docking results revealed that IPM714 may inhibit the P13K/AKT/mTOR pathway, which controls the cell cycle and apoptosis, to reduce cell proliferation. IPM714 is a promising medicine for the treatment of colorectal cancer, according to the study [33-35].
Newly created inhibitors of tubulin polymerization and anticancer drugs called imidazole chalcone derivatives (9) (Figure 3) [36, 37]. On a number of human cancer cell lines, including A549 (adenocarcinoma human alveolar basal epithelial cells), MCF-7 (human breast cancer cells), MCF-7/MX (mitoxantrone resistant human breast cancer cells), and HEPG2, they examined the antiproliferative effect of imidazole-chalcone (human hepatocellular carcinoma cells). Compared to the other three, the imidazole-chalcone derivatives generally exhibited more cytotoxicity against A549 cancer cells. According to the flow cytometry analysis, these compounds produced cell cycle arrest at the G2/M phase at low doses but increased the number of apoptotic cells in higher concentrations. The molecular docking investigations of 6455 into the tubulin colchicine binding site suggested that this substance may interact with tubulin.

Figure 3.
Based on the hybrid pharmacophore method, new imidazole-1, 2, 3-triazole hybrids (10) were created and produced (Figure 4) [38-40]. Both elemental analysis and several spectrum approaches are used to describe the substances. Using the MTT assay, the synthesised compounds were tested for their anticancer activity against the Caco-2, HCT-116, HeLa, and MCF-7 cell lines. The substances demonstrated strong antitumor action. The results of in silico molecular docking experiments using ADMET profiles and glycogen synthase kinase-3 are evaluated.

Figure 4.
A number of new imidazole derivatives with benzene sulfonamide moiety in their structure were created [41, 42]. Using the MTT test on the human triple-negative breast cancer MDA-MB-231 and the human malignant melanoma IGR39 cell lines, the cytotoxicity of generated compounds is evaluated. According to the results of the assay, the chemicals were more effective against the MDA-MB-231 cell line used to model human triple-negative breast cancer. The compounds that contained benzene sulfonamide and imidazole derivatives with 4-chloro (12) and 3, 4-dichlorosubstituents in the benzene ring and 2-ethylthio (12) and 3-ethyl (11) groups in the imidazole ring were the most active ones (Figure 5).

Figure 5.
A novel series of bis-imidazoles and bis-imidazo[1,2-a]pyridines (13) from Schiff’s base dimers for their anticancer activities were designed and synthesized (Figure 6) [43, 44].

Figure 6.
Three cancer cell lines, including cervical (HeLa), breast (MDA-MB-231), and renal cancer, were used to test the synthesised compounds for anticancer activity (ACHN). Studies on the connection between structure and activity revealed that imidazo[1,2-a]pyridines have outstanding cytotoxic properties. The substance displayed below exhibited the strongest inhibitory effects on all three cell lines, particularly the breast cancer cell line. These chemicals produced in vivo anticancer effects that were comparable to those seen in mice given tamoxifen as a routine treatment.
Derivatives of dehydroabietylamine (14), (15), and (16) were created (Figure 7), and the MTT assay was used to test their anti-cancer effects on HeLa (cervical), MCF-7 (breast), A549 (lung), HepG2 (liver), and HUVECs (umbilical vein, normal cells) in vitro [45]. The findings demonstrated that almost all of the compounds had improved anti-cancer activity (Table 1).

Figure 7.
Table 1. Cytotoxicity of compounds against cancer and normal cells (IC50 ± SE /μ M )

Developed new N-(6-substituted-benzothiazol-2-yl), the cytotoxic effects on C6 and HepG2 tumour cells of -2-[4, 5-dimethyl-1-((p-tolyl/4-nitrophenyl)amino)-1H-imidazol-2-yl] thio] acetamide derivatives (17) were evaluated for their anticancer activity (Figure 8) [46].

Figure 8.
The novel imidazo[1,2-a][1,3,5]triazines were synthesized in a series, and their derivatives (18) were discovered (Figure 9) to be selective FAK inhibitors against the tested kinases [47-49]. The IC50 values for the compounds were 10-7-10-8 M. Some of those substances effectively slowed the growth of a group of cancer cell lines that expressed a lot of FAK. Apoptosis study in U87-MG and HCT-116 was performed, and the results showed that these chemicals slowed down cell growth by halting cells in the G2/M phase of the cell cycle. The substances severely reduced U87-MG cells' ability to adhere to, move through, and invade the cell matrix.

Figure 9.
2.2. Antioxidant Activity
Based on a number of studies, investigations showed compounds containing imidazoles had intriguing antioxidant properties [50]. This section highlights some developments in the realm of antioxidant chemicals based on imidazoles.
Functional arylimidazole derivatives modified with polyphenol residues like phloroglucinol, pyrogallol, and hydroxyquinol were explored [51, 52]. A number of 5-aryl-2H-imidazole family bi-functional chemical compounds (19) were explored (Figure 10). The bi-functional 5-aryl-2H-imidazoles' structure and antioxidant characteristics were investigated. To gauge the antioxidant capabilities, they used the AOC, ARC, Folin, and DPPH tests. The outcomes demonstrated that the compounds family under consideration can display the antioxidant action.
Utilizing the 1,1-diphenyl-2-picrylhydrazyl (DPPH) and 2-2′azino-bis-(3-ethylbenzothiazoline-6-sulfonate) (ABTS+) tests, a series of 2,4,5 triphenyl imidazole derivatives were created, described, and evaluated as antioxidant agents in vitro [53]. Additional in silico research, including docking, was carried out. In addition, the parameters of absorption, distribution, metabolism, and excretion were calculated. Two of the synthesized compounds shown below were the most effective antioxidants in the DPPH and ABTS assays, with ECs of 0.141(20) and 0.174 mg/ml (21), respectively, and 0.168 and 0.162 mg/ml (Figure 10).

Figure 10.
A series of naphthalene incorporated 2, 4, 5-trisubstituted imidazole derivatives (22), (23), and (24) were synthesized by one pot multicomponent reaction and characterized by FT-IR, N-MR, and mass spectral analysis [54, 55]. The final compounds were investigated for their antioxidant activities. The biological study of synthesized compounds showed the promising antioxidant activity (Figure 11).

Figure 11. Naphthalene incorporated 2, 4, 5-trisubstituted imidazole derivatives
Using the Radziszewski reaction, a novel series of tri-substituted imidazole derivatives were produced [56, 57]. The chemicals were created through a reaction between benzil, ammonium acetate, and several 1H-pyrazole-4-carbaldehyde derivatives. Spectral analysis and elemental analysis (CHN) were used to confirm the structures of produced compounds (FTIR, 1H, and 13C-NMR, and LC-MS). The DPPH technique was used to test the synthesised compounds (25) for their antioxidant properties. The substances displayed anti-DPPH radical antioxidant action (Figure 12).

Figure 12.
2.3. Antibacterial activity
Since bacterial drug resistance is the primary factor for many antibiotics to become less effective, it is a public health concern. Therefore, it is crucial to create new and more effective antibacterial agents. Imidazole and its derivatives have been shown by many researches to have remarkable antibacterial action.
By reacting ammonium acetate, substituted aldehydes, and benzil with tartaric acid as an organocatalyst, three components-ammonium acetate, substituted aldehydes, and benzil-were able to be synthesised in a multi-component process to create imidazole derivatives [58]. The synthesised compounds (26) (Table 2) have also been tested for their antibacterial activities against Salmonella typhii, Escherichia coli, Bacillus subtilis, and Pseudomonas aeruginosa by disc diffusion method using gentamicin as a reference and DMSO as a negative control. Molecular docking studies have been conducted against the bacterial protein GlcN-6-P and obtained conformations of ligand complexes and proteins. When used against Salmonella typhii, Escherichia coli, and Bacillus subtilis, the imidazole derivatives had potent antibacterial action. However, when used against Pseudomonas aeruginosa, it had only moderate antibacterial activity (Table 3).
Table 2.

Table 3. Studies of trisubstituted imidazole derivatives against bacterial protein using molecular docking
N-(4-((benzyl)oxy)phenyl) acetamide derivatives of 2-((5-acetyl-1-(phenyl)-4-methyl-1H-imidazol-2-yl)thio) were developed and created [59]. The conjugates have significant antibacterial activity against both gram-positive and gram-negative bacteria according to tests on antibacterial activities. In addition, a number of derivatives were tested for their ability to inhibit in vitro resistant bacterial strains like methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococcus, and extended spectrum beta lactamases (ESBL) (MRSA). Studies using the Lipinski rule of five and ADME were carried out on substances containing the Staphylococcus aureus dihydropteroate synthase (saDHPS) protein (PDB ID:6CLV).
Novel 1-(furan-2-yl)methyl)-4,5-diphenyl-2-p-tolyl-1H-imidazole (FMDI) molecules have been created [60]. These compounds' structures were validated by contemporary techniques such as single crystal XRD, 1H-, and 13C-NMR as well as IR. Density functional theory was used to investigate the compound's shape. Using natural bond orbital analysis, it was possible to study the stability and charge delocalization of the molecule. The charge transfer that occurs inside the molecule is described by the HOMO-LUMO energies. The parameters of thermodynamics are calculated, and their values rise as temperature rises because vibrational intensities increase. Utilizing molecular docking experiments, the antibacterial properties of these compounds were investigated. According to molecular docking experiments, the substance has higher binding energies than Mitomycin and the common antibiotic Ciprofloxacin.
Binding energy: -11.69kcal/mol and 15.40kcal/mol when compared to standard drug Ciprofloxacin and Mitomycin.
4, 5-diphenyl-N-alkyl imidazole derivatives (27) were created, and their antibacterial activity was assessed (Figure 13) [61]. Spectroscopical methods validated the chemicals. S. aureus, B. subtilis, and E. coli, and the examined bacteria have only the moderate action against chemicals.

Figure 13.
A few fresh imidazole analogues were created, and their anti-HIV activity was assessed [62]. According to the findings, a variety of imidazole and nitro imidazole analogues exhibited a modest level of antibacterial activity. In accordance with the findings, N-(3-hydroxyphenyl)-2-(substituted imidazole-1-yl) alkanamides also displayed noteworthy antibacterial activity.
2.4. Anti-tubercular activity
Mycobacterium tuberculosis, which causes TB, is an infectious disease that typically affects our lungs. The biological actions of imidazole and its derivatives are diverse, and several of them have shown the excellent anti-tubercular activity.
Researchers produced 2, 4, 5-trisubstituted imidazole derivatives in a single pot using benzil, ammonium acetate, and 2-phenoxyquinoline-3-carbaldehyde as starting materials and ceric ammonium citrate as the catalyst [63, 64]. Using mass spectroscopy, IR, 1H-NMR, 13C-NMR, and 1H-NMR to confirm the compounds' structures, the anti-tubercular activity of the produced compounds (28) was tested (Figure 14).

Figure 14.
The novel pyrido[1,2-a]imidazo-chalcones have been synthesised, and they have been tested for their anti-tubercular activity against the Mycobacterium TB H37Rv strain as well as their cytotoxicity in Vero cells (C1008) and mouse bone marrow derived macrophages (MBMDM) [65, 66]. The following substances were discovered to be the most active of the investigated substances, with MIC values of 7.89 (29), 6.42 (30), and 6.59 (31) M, respectively (Figure 15).

Figure 15. Pyrido[1,2-a]imidazo-chalcones
2-t-Butyl-4, 5-diarylimidazoles (32) were created and put into production, and they were discovered to be MtGS inhibitors with submicromolar IC50 values and hopeful antituberculosis MIC values (Figure 16) [67-70].

Figure 16.
2.5. Anti-Inflammatory activity
Non-steroidal anti-inflammatory medications are prescribed to treat inflammatory conditions such as fever, discomfort, and inflammation. Inhibiting the enzyme cyclo-oxygenase (COX), which transforms arachidonic acid into prostaglandins, thromboxanes, and thromboxanes, is the principal mechanism by which they work [71]. Imidazole-containing compounds demonstrated considerable anti-inflammatory effect among the other biological properties.
Various reaction conditions were used to create a number of imidazole derivatives from 2-(4-chlorophenyl)-4, 5-diphenyl-1H-imidazole-1-yl)-acetic acid hydrazide. Elemental analysis, IR, H NMR, and mass spectral data were used to confirm the compounds' structural details [72]. All the synthetic compounds (33) had moderate to good anti-inflammatory activity (Figure 17).

Figure 17.
Epiisopiloturine (34), an imidazole alkaloid present in Pilocarpus microphyllus leaves, examined for its anti-inflammatory properties (Figure 18) [73-75]. Several substances that cause Swiss mice to develop peritonitis and paw edoema are used to test the compound's anti-inflammatory effects.

Figure 18. Epiisopiloturine
2.6. Antiviral activity
Nowadays, viral infections are on the rise, making it crucial to create new antiviral medications with improved action and less resistance. Antiviral medication development is a difficult procedure because antiviral medications prevent the growth of their intended pathogens rather than destroying them. It is challenging to develop a safe and effective antiviral medicine since viruses require the host's cells for replication. Numerous investigations have demonstrated the antiviral action of imidazole nuclei. An updated overview of imidazole analogues with antiviral activity is provided in this section.
Condensation reaction of azo linked ortho-vaniline precursor with amino functionalized imidazole derivatives produced four novel ionic liquid tagged azo-azomethine derivatives [76]. Different analytical and spectroscopic methods were used to characterise the synthetically produced compounds. They used molecular docking studies to determine whether the examined ligands had an inhibiting effect on the primary protease (6LU7) of new coronaviruses (COVID-19).The results of docking studies showed that there is a significant inhibitory action against the main protease of SARS-CoV-2. The binding energy values of the four ligands against the protein 6LU7 have found to be -7.7 Kcal/mole (35), -7.0 Kcal/mole (36),-7.9 Kcal/mole (37), and -7.9 Kcal/mole (38) (Figure 19). The efficiency of ligands was compared with the FDA-approved drugs such as remdesivir, chloroquine, and hydroxychloroquine. From the comparison, it was concluded that the ligands could act as an inhibitor against SARC-CoV-2. Pharmacokinetic properties were also studied which revealed that ligands (35-38) act as potential drug candidates.

Figure 19.
Synthetic 1-hydroxy-2-(2-hydroxyphenyl) imidazole derivatives (39) were tested in Vero cell culture for their antiviral potency against the vaccinia virus (Figure 20) [77-80]. They discovered that some 1-hydroxyimidazole compounds exhibited the modest cytotoxicity and antiviral activity.

Figure 20.
From derivatives of imidazole and coumarin, a variety of new conjugated compounds with a -SCH2 bond were produced via chemical methods [81-84]. According to the data, three of the recently created imidazole coumarin conjugates (40), (41), and (42) have attractive EC50 values (Figure 21) [5.1-8.4 M] and selective indices >20 against the hepatitis C virus. A hydrogen atom at the N1 position in the imidazole nucleus and a substituent, such as Cl, F, Br, Me, or OMe, in the coumarin nucleus were added to the structure to boost the potency and selectivity.

Figure 21.
Imidazole-4, 5-dicarboxamide derivatives that target the dengue fever virus were created and tested [85, 86]. The imidazole-4, 5-dicarboxamide derivative (43) has a strong dengue virus inhibitory activity, according to the results of a high throughput screening experiment using the dengue virus-2 replicon.
4-(Phenylcarbamoyl)-1H- imidazole-5-carboxylic acid was discovered as a potent and selective inhibitor of the in vitro interaction between HIV-1 integrase and LEDGF/p75, which they subsequently used to help create novel allosteric inhibitors [87]. A family of 5-carbonyl-1H-imidazole-4-carboxamides (44) that can inhibit the HIV-1 integrase-LEDGF/p75 interaction in vitro was produced as a result of a chemical synthesis guided by molecular docking (Figure 22).

Figure 22.
2.7. Antifungal activity
34 imidazole-based compounds were created using a one-pot catalytic process, and their antifungal efficacy was tested against a variety of fungi [88-91]. The interesting antifungal activity was seen in some compounds, both strongly and moderately. Studies on molecular docking were conducted. The two most active compound’s (45) (IC50 - 95±7.07μ M) and (46) (IC50- 235±7.07μ M) docking tests revealed that they might function by preventing the fungus' lanosterol 14-methylase from doing its job (Figure 23).
Figure 23.
The antifungal activity of a new series of 2-(1H-imidazol-1-yl)-1-phenylethanol aromatic ester and carbamate derivatives was examined against strains of Candida albicans and non-albicans Candida species [92]. The aromatic biphenyl ester derivatives of the compound were discovered to be more effective than the reference substance. To obtain pure enantiomers, the compounds' racemic mixtures underwent purification. It was discovered that (+) isomers were 500 times less active than (-) isomers.
2.8. Anti-parasitic activity
Various studies showed evidence that supports the antiparasitic action of imidazole derivatives.
A number of novel imidazole derivatives, including phenyl-substituted 1H-imidazoles (47), (48), thiophene imidazoles (49), and bis-imidazoles were created (Figure 24) [93-95]. The ability of the substances to inhibit Toxoplasma gondii development in vitro was evaluated. Studies were conducted to predict bioactivity and conduct molecular analysis. The outcomes demonstrated that imidazole derivatives are highly selective against T. gondii as opposed to the host cells. Some imidazole derivatives among the evaluated compounds shared a particular structural component and demonstrated noticeably strong selectivity towards the parasite vs. the host cells.
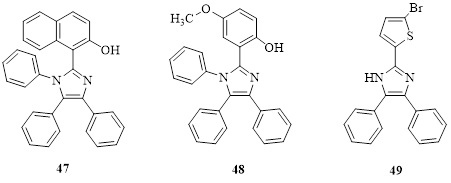
Figure 24.
To treat parasite disorders like Leishmaniasis, Chagas disease (Trypanosoma cruzi), and Sleeping sickness (Trypanosoma brucei), new medications should be quickly discovered and developed (Leishmania ssp) [96, 97]. According to the World Health Organization (WHO), these illnesses are among the top thirteen most neglected diseases in the world. The synthesis of 14 aryl substituted imidazoles (Table 4) and their molecular docking onto sterol 14-demethylase (CYP51) were carried out in the current work. The antiprotozoal activity of substances against T. cruzi, T. brucei, T. b. rhodesiense, and L. infantum was further assessed. All samples from aryl substituted imidazole compounds displayed intriguing antiparasitic activity to vary degrees, according to in vitro antiparasitic results of the aryl substituted imidazoles against T. brucei, T. cruzi, T. b. rhodesiense, and L. infantum (Table 5). Strong binding was shown by the ligands 50a, 50c, 50e, 50f, 50g, 50i, and 50j. Only the ligand 50c shown good selectivity against all examined parasites while the majority of compounds were cytotoxic to MRC-5 cell lines (1.12< CC50 <51.09μ M). According to the molecular docking data, the oxygen from NO2, SO3H, and OH groups interacts with the Fe2+ ion of the heme group, whereas the aromatic substituents in positions 1, 4, and 5 have primarily stabilising hydrophobic interactions with the enzyme matrix.

Figure 25.
Table 4.

Table 5. Antitrypanosomal, antileishmanial (IC50, μM) and cytotoxic (MRC-5) (CC50 μ M) activities of arylsubstituted imidazoles.

- Conclusion
According to the current review, imidazole derivatives have beneficial pharmacological properties such as anti-tubercular, anti-cancer, antioxidant, antibacterial, antifungal, antiviral, and antiparasitic activity with a great potential to treat various human diseases. Our analysis of the findings from numerous studies revealed that a number of imidazole derivatives have a tolerable pharmacokinetic profile and a promising bioavailability score. To put it briefly, the current reviewed research on imidazole scaffold suggests further development and optimization to find new and effective therapeutic candidates.
Acknowledgment
Grateful thanks to Dr. Vinod B for his guidance with the task. I want to sincerely thank my colleagues especially the students of Masters in Pharmaceutical Chemistry for their grateful support.
Orcid:
R. Lakshmidevi: https://orcid.org/0000-0001-7635-9130
Das Reeja: https://orcid.org/0009-0007-8778-1864
Rohith Rajan: https://orcid.org/0009-0001-5053-9982
Vinod: https://orcid.org/0000-0002-90510412
Citation: V. R. Lakshmidevi*, D. Reeja, A. Rohith Rajan, B. Vinod, Advanced Spectrum of Imidazole Derivatives in Therapeutics: A Review. J. Chem. Rev., 2023, 5(3), 241-262.
)