Document Type : Review Article
Authors
- Babafakruddin Peddagundam
- Hindustan Abdul Ahad
- Haranath Chinthaginjala
- Tarun Ksheerasagare
- Aravind Kumar Ganthala
- Govardhan Reddy Gummadisani
Department of Industrial Pharmacy, Raghavendra Institute of Pharmaceutical Education and Research (RIPER)-Autonomous, K. R. Palli Cross, Ananthapuramu-515721, Andhra Pradesh, India
Abstract
This work explores soft drugs and their configuration for drug delivery. Compared with the stimulated soft compound, soft drugs have a fast metabolism and exert their therapeutic effects. They are organically active drugs, carefully formulated to have a predictable and controllable absorption near harmless compared with sedentary products. The broader neurometabolic drug plan concept depends on the soft drug configuration. This is usually accomplished by using close analogues of proven lead compounds and an ester as a metabolically delicate weakness. A rationally designed or accidentally developed soft drug has proven clinically useful. The idea of neurometabolic drug plans relies upon the configuration of soft drugs. This method is immensely flexible and can be implemented to create inventive, new substances in a widespread helpful field. A metabolically, especially hydrolytically labile moiety, is attached to the construction to control and direct digestion.
Graphical Abstract
)
Keywords
Main Subjects
- Introduction
Soft Drugs (SDs) are organically active drugs, careful to take a foreseeable and controllable absorption near harmless besides sedentary foodstuffs. Consequently, they consume their anticipated pharmacological consequence[1]. The molecule could stay disabled, and then be disinfected shortly after it has drilled its biological effect [2]. The therapeutic index could be amplified, providing a safer drug. SDs are not supposed to be the basis for physical or psychological need or addiction to the number of hard drugs, but they are immobile and dangerous. Samples of soft drugs are hallucinogens like cannabis[3], mescaline [4], psilocybin [5], Lysergic acid diethylamide (LSD) [6], ayahuasca [7], iboga [8], and dimethyltryptamine (Figure1) [9]. Even though they do not source animal habits, some of them may be immovable due to psychological requirements. There is some ominous omen about hallucinogens calming customs nearby. Given that medicines are not perfectly prepared as correspondingly soft or hard drugs and have physiognomies of both. Examples of such drugs are 3,4-Methylenedioxy Methamphetamine [10], ketamine [11], Phenyl cyclohexyl Piperidine (PCP) [12], dextromethorphan (DXM) [13], synthetic cannabis, and caffeine [14].
Consequently, these canisters remain definite as medicines which remain crop predictable and manageable in vivo metabolism to form nontoxic crops. Consequently, they consume them, exposing their sustaining role [15] among which acetyl pyridinium chlorides, soft chloramine are mentioned.

Figure 1. a) Mecalin structure b) LSD structure c) Iboga structure
- Pharmaceutical application of prodrugs to elivate patient satisfactoriness
An model prodrug is a organic thing which has no pharmacological motion contrary to a nominated physical goal, but is metabolically altered addicted to a compound through the anticipated motion. Prodrugs can be exploited for a variability of resolutions, together with enhancing bioavailability or pharmacokinetics of a medicine, diminishing medication harmfulness, simplifying management of the medication, or transporting the medication to detailed cells or tissues. Approximately medicines, such as the antituberculosis agents isoniazid and ethionamide, consume before initiate to remain prodrugs.
2.1 Taste improvement
The deliciousness of the energetic component of a medication is a substantial hindrance in emerging a patient approachable quantity arrangement. Around remain plentiful pharmaceutical and over the counter (OTC) preparations which hold energetic elements, which stay nasty in sense of taste. Regarding OTC preparations, by means of cough and cold syrups, the unpleasantness of the preparation remains to the absence of patient acquiescence.
- Drugs with unpalatable taste [16],
- Drugs which are unsuitable for preparation as a suspension [17],
- Diminished drug solubility in the saliva [18].
Here, some instances for parent drug and its prodrug with improved taste (Figures 2, 3 and 4)[18-20]:

Figure 2. Conversion of Chloramphenicol Palmitate to Chloramphenicol [21]

Figure 3. Conversion of Clindamycin Palmitate to Clindamycin [22]

Figure 4. Conversion of Sulfafurazole to Diacetate ester [18]
2.2 Feature
It has a close structural similarity to the lead, and also has a metabolically sensitive moiety built into the lead structure. The incorporated metabolically sensitive spot does not affect the overall physicochemical or steric properties of the lead compound.
- It consumes a nearby organizational mainly compared with the chief.
- It consumes a metabolically created delicate moiety to be enraged by the leading structure.
- The combined metabolically delicate advertisement does not touch the perfect physicochemical and steric belongings of the main complex [23].
2.3 Advantages
Some merits of soft drugs are mentioned as follow [24, 25]:
- The drug’s therapeutic index rises.
- Evasion of pharmacologically active metabolites which can progress in to long-term effects.
- Elimination of drug interactions caused by enzyme metabolite reserves.
- Interpretation of pharmacokinetic problems caused by numerous dynamic species.
- The altered prodrugs are further soft drugs.
- The perceptions of prodrugs and soft drugs remain different.
- A prodrug is an inactive composite which involves metabolic adaptation to the energetic method.
- A soft drug is pharmacologically energetic, and then habits break down as per an income of encouraging defecation.
- Organization of soft drugs
Soft drugs are the isosteric or isoelectronic analogs of a lead compound which have a predictable and controllable metabolic route so that the drug’s toxic effects could be overcome with an improvement in the therapeutic index of the drug. The desired activity of the soft drugs is generally local and they are applied on or near the site of action. Thus, the soft drugs produce their pharmacological effect locally, but their distribution away from the site of action results in metabolic deactivation, which avoids undesirable activity and toxicity.
Soft drugs are unglued through Bodor hooked on five dissimilar assemblies [26, 27].
3.1 Soft analogues
Soft analogues are close structural analogues of known active drugs or bio active compounds. These compounds have specific metabolically sensitive spot built into their structure which provide their one-step controllable detoxification. These sensitive spots are not oxidizable alkyl chains or functions groups subjected to conjugation. The designed detoxification will take place as soon as possible after the desired activity is achieved.
- Soft similarities remain near mechanical similarities of recognized energetic drugs before bioactive complexes [28].
- These complexes consume a detailed “metabolically penetrating advertisement” manufactured into their construction, which affords their one-step controllable detoxification [29].
- Before functional groups are exposed near these sensitive spots, they remain non-oxidizable alkyl cuffs. The intended cleansing occurs aggressively later of its activity seized [30].
- The simplest sample of the soft equivalent remains the isosteric analog (II) of cetylpyridinium chloride (I), which is a solid quaternary antimicrobial agent [31].
3.2 Simulated soft compounds
The simulated soft compounds imitating the conditions of something especially as a training the ecercise of cetylpridinium chloride and non-oxidizable.
- These complexes remain the equivalents of identified drugs [32].
- These remain considerably through presenting a pharmacophoric assembly to a harmless sedentary complex to stimulate it in order to exhibit a positive pharmacological movement [33].
- In vivo, the activated form determination loses the activating group and reverts to the original non-toxic compound [34].
- For instance, soft chloramine is less acidic (anywhere the chlorine atom is involved with a heteroatom) than the conservative chloramines. For example, chloramine-T is accessible in salt form, so it stays less corrosive [35].
3.3 Normal soft drugs
Endogenous materials can be regarded to be the ordinary soft drugs, and the body possesses efficient, fast metabolic trails for their statement without going through extremely reactive intermediates [36].
For example, neurotransmitters and steroidal hormones [37].
For instance, the use of diesters of adrenalone to distribute the epinephrine to the judgment through joint reduction after hydrolysis process [38].
3.4 Soft Drugs founded proceeding energetic metabolite method
Soft drugs are therapeutically active compounds which undergo a predicated fast metabolism into inactive metabolites after exerting their desired therapeutic effects of soft drugs design as to control and direct metabolism typically by incur-proration of a metabolically sensitive moiety into the structure.
- Some drugs undergo stepwise biotransformation into philanthropic intermediates and organizational equivalents through which movement is compared with that of the innovative particles [39].
- According to Bodor, it is desired to use it as a high quality drug and its active type which undergoes a one-step, singular, and predictable metabolic deactivation [40].
- Oxyphenbutazone is the active p-hydroxy metabolite of phenylbutazone [41].
- Oxazepam is the public dynamic metabolite of chlordiazepoxide, halazepam, chlorazepoxide, and diazepam [42].
3.5 Soft drugs based arranged sedentary metabolite method
This is one of the most useful and successful strategies for designing safe and selective soft analogues. Likewise, it involves the metabolism of active species which deals with the structural modifications in the inactive, excreted metabolite of an active drug in order to allow the metabolic reconversions to occur in a facile, one step, and controllable manner, with a return to the very inactive metabolite from which the design began.
This is done in three pahses which are mentioned as follow [43, 44]:
- Stimulation phase: This is a biological variation of an acknowledged “inactive metabolite of a medicine (through iso asterism). For instance, this metabolite is cast-off, as per the main compound [45].
- Predictable metabolism: Such a method is popular, because its metabolism determination produces the preliminary inactive metabolite in a single step without the use of toxic intermediates [46].
- Manageable metabolism: Managing transport and binding properties as fine as possible in a place of metabolic rate and pharmacokinetics in a molecular modification [47].
Chlofenotane, the acid metabolite “v” which remains inactive with comparatively squat toxicity excreted as water-soluble species, is a main composite for the inactive metabolite method. That remains the ethyl ester of clofenotane.
- Soft drug design-basic principles
The SD thought is a part of the extra general gratitude which drug design needs to fully integrate metabolic thoughts from the very beginning, as metabolites subsidize meaningfully to the overall motion and toxicity of the drug level, not on enlightening activity deprived of help, however on refining the activity/toxicity ratio. This is frequently characterized by the therapeutic index, typically defined as the percentage between the half-maximal toxic and effective doses: TI = TD50/ED50. These ideas are the foremost underlying principles of retro metabolic drug design, which incorporates both SD and chemical delivery system (CDS) design [48].
This container leads to intricate time profiles as per proved in which the event of a hypothetical drug D is composed of its energetic, poisonous, and sedentary metabolites. In roughly 70% of poisonous medicines, sensitive metabolite production remains a cause of poisonousness. A well-designed SD container leads to an abundant and more basic scenario. For SDS, inactivation should stay comparatively dissolute, and then be permitted to interfere with imaginable drug-drug communications. As cytochrome P450 enzymes, such as, for instance, CYP-3A, 2C9, and 2C19 oxygenases which remain in control used for the breakdown of most drugs are saturable and subject to inhibition and induction, inactivation by hydrolytic enzymes could be an improved high-quality. This absorption (e.g., cleavage of an ester bond) can be passed out quickly and even extrahepatically through universally dispersed esterases, which possibly will not remain free of embarrassment or polymorphism issues, far less CYPs (e.g., there is no known carboxylesterase-mediated drug) [49].
Drug connections popular at the clinic are so far away which they are used only for about-stated communications through ethanol. Hydrolytic degradation also consumes the gain which its procedures give an acidic metabolite, which then announces a harmful responsibility that it is possible to not fit sufficiently into the ligand-binding site, significantly decreasing the receptor necessary for the innovative ester drug and elevating the chance that the metabolite is certainly indolent. Despite the existence of theoretical converses, SDs remain occasionally immobile jumbled through prodrugs, mostly for both experiencing projected metabolic variations and together, mostly proceeding enzymatic hydrolysis. SDS, though, is dynamic and remains deactivated by an in-built mechanism. However, prodrugs are sedentary and must be stimulated. A significant portion of the current small-molecule drugs (10–15%) are, indeed, prodrugs. Most, in contrast, are chance prodrugs which involve metabolic beginnings, then remain unintentionally considered to do so. Balanced prodrug design, on the other hand, is progressively positive and has resulted in around 30 clinically approved products, accounting for 10% of new drugs in the last decade. Inappropriately, the SD/prodrug mix-up is only dependent on authors who use the antidrug term to designate SDs. This is deceptive and must continue to the end [50].
An anti-drug proposes the same thing as a prodrug (i.e. a requirement used for metabolic stimulation rather than inactivation), because the Latin ante-prefix has a similar denotation to the Greek pro- (e.g., before, precedent).
The effects design of metabolites impacts on the overall effects and toxicity of therapeutic agents. In the case of a hypothetical drug which is metabolized into energetic, poisonous, and inactive metabolites (Ma, Mt, and Mi, respectively), corresponding time profiles for absorptions, effects, and harmfulness (arbitrary units) are exposed for all mechanisms presumptuous which D is broken down at dissimilar rates into these metabolites, and they all consume dissimilar removal toxins as well as actions and poisonousness. As per this issue, most of the effect is donated by the parent drug (D) and its active metabolite Ma, but the toxic metabolites make the problem (Mt). The term “equivalent” is meant for an assumed soft drug (SD) which is absorbed rapidly into a sedentary metabolite (Mi) of negligible movement and harmfulness. As opposed to A, both activity and toxicity are determined only by the people of the innovative SD, even if Mi absorptions are quite high. Hence, complex PK profiles and poisonousness due to responsive metabolites are evaded, and movement and toxicity can be better regulated. Time outlines were obtained using single-compartment representations with interest, metabolism, and elimination rate constants (kabs, km, a, kel, etc.) and consistent discrepancy equations as specified. Toxins and belongings were assumed to be continuously relative to absorptions (i.e. there was no effect compartment) [51].
Following management, numerous metabolites are generated for most medications, and these can have a substantial impact on not only overall activity, but also on toxicity and adverse effects. As seen in the displayed case of a hypothetical drug (D) and its dynamic, toxic, and sedentary metabolites, this container leads to complex temporal profiles. In over 70% of drugs connected to toxicity, the development of sensitive metabolites remained a cause of poisoning. A stylish SD can be the center of attention in a more basic situation [52].
For SDS, inactivation had better be comparatively dissolute and allow for interference from imaginable drug-drug interactions. Since cytochrome P450 enzymes, (CYP3A, CYP2C9, and CYP2C19 oxygenases which are accountable for the metabolism of most drugs as being saturated and subject to the self-consciousness and induction, inactivation by hydrolytic enzymes could be an improved choice. This metabolism (e.g., cleavage of an ester bond) can be approved out quickly and even extrahepatically by widely dispersed esterases, which may not be perfectly free from replacement or polymorphism issues, but are far less than CYPs (e.g., there are no recognized carboxylesterase-mediated drug-drug communications in the clinic except for a few reported relationships with ethanol). Hydrolytic withdrawal has the advantage of producing an acidic metabolite with a negative charge which is unlikely to fit into the ligand-binding site, weakening receptor binding significantly compared with the original ester-containing drug and increasing the likelihood that the metabolite is inactive [53].
Despite life’s theoretical reverses, SDs remain occasionally unmoving and disordered through prodrugs, mostly to experience projected metabolic variations together, then trusting mostly in enzymatic hydrolysis. Prodrugs remain active and deactivated through an integral device in which prodrugs remain sedentary and the requirement remains stimulated. An important percentage of current small-molecule medicines (10–15%) are, in fact, prodrugs. Most, though, are involuntary prodrugs which need metabolic stimulation, and therefore they were not purposely considered to be prepared. However, the prodrug enterprise is becoming more effective as a source of some clinically accepted products, accounting for 10% of new drugs approved in the last decade. Inopportunely, the SD/prodrug muddle is only extended by some novelists who use the antidrug term to designate SDs. Because the Latin ante-prefix is similar to the Greek pro- (e.g., before, precedent), anti-drug implies the same thing as a prodrug (i.e. the requirement used for metabolic stimulation rather than inactivation) and is thus the inverse of what it is theoretically used [54].
- Clinical success stories
The proposed study is mainly a type of research study which tests the applications of the modern medical approaches. These studies test new methods of screening, prevention, diagnosis, or treatment of a disease. In the four decades since the summary of the concept, the SD method has remained popular and nearly altogether evolving in several hypothetical manufacturing investigation hubs, including some main therapeutic corporations some of which are controlled by clinically recognized products [55] as sensibly designed SDs which, for the case, stretch marketing approval boundaries.
- The ultra-short-blockers esmolol and landiolol are forgiving-blockers.
- The forgiving glucocorticoid loteprednol etabonate is accepted for seditious and allergy-related ophthalmic syndromes.
- The ultra-short-acting soft opioid palliative remifentanil.
- Remimazolam, a soft benzodiazepine is accepted for universal anesthesia.
- The ultrashort-acting soft calcium-channel blocker clevidipine is accepted for using as the saving controller of plasma compression in cardiac clinical procedures.
Rationally designed or accidental (acid) soft drugs which are accepted for clinical use (provided trademark) or have made clinical progress. For each compound, the entity responsible for its development, the year of the first journal, and the appropriate trademark (unknown accessible) are also included [56]. The approved soft drug details were illustrated in (Table 1).
Table 1. Approved soft drug compounds

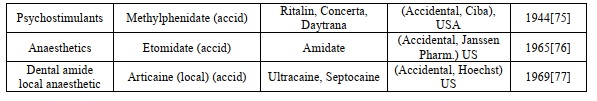
The abbreviation list is illustrated in Table 2.
Table 2. List of abbreviations used in the manuscript

- Conclusions
Soft drug configuration is significant for the broader neurometabolic drug plan idea. It is exceptionally broad and can be applied in a wide scope of helpful regions to create inventive and new substance elements. The objective is to control and direct digestion by a fuse of a metabolically, especially hydrolytically labile moiety into the construction. As a rule, this is accomplished by means of SDs which are close analogues of realized fruitful lead compounds and by involving an ester as the metabolically delicate weakness. Noteworthy, both again plan (for instance, the novel underlying systems) and non-ester type weaknesses are conceivable. All SD projects need to incorporate nitty gritty metabolic portrayal and affirmation of the latency of the metabolite. SDs are especially appropriate for helpful applications where the ideal action is confined, short or ultrashort, or powerless to simple titration. Therefore, anaesthesiology, dermatology, ophthalmology, and inward breath therapeutics address areas of specific guarantee. During the forty years since the presentation of the idea, a few objectively planned SDs got advertising endorsement.
)