Document Type : Review Article
Authors
1 Research Institute of Medicinal and Aromatic Plants (RIMAP), Beni-Suef University, Egypt
2 Basic sciences department, Higher Technological Insitute-Beni Suef, Egypt
3 Chemistry department, Faculty of science, Beni-Suef University, Egypt
4 Micro-Analysis, Environmental Research and Community Afairs Center (MAESC), Faculty of Science, Beni-Suef University, Beni-Suef, Egypt
Abstract
In this study, we summarized the preparation, characterization, and computational research on triazolo pyrimidine derivatives utilizing the Density Functional Theory technique. Quantum mechanics calculations and thermodynamic parameters show that energy exchange takes place within molecules. Geometrical and structural aspects such as dipole moment, relative populations, relative total energies, electronic total energies, vertical emission energies, bond length, and bond angle were also discussed in this study. The triazolo pyrimidine ring is a structural characteristic found in a variety of active molecules with varying pharmacological activity. During the last few decades, a vast amount of published literature has been reviewed. This review covers numerous triazolo pyrimidine preparations, characterizations, and computational analyses, and it might be considered the lead compound for future medicinal and agrochemical development.
Graphical Abstract
)
Keywords
Main Subjects
- Introduction
Triazolo pyrimidine derivatives are fused heterocyclic compounds which have found use in a range of industries [1-4]. Triazolo pyrimidines, 1,2,4-and 1,2,3-triazolopyrimidines, are utilized in anti-cancer [5-7] , anti-tumor [8], anti-inflammatory [9], anti-bacterial [10, 11], anti-fungal [12], and anti-malarial [13] therapeutic and pharmacological uses[6], [14-16]. They are anti-Leishmania [17, 18], anti-viral [19, 20], anti-HIV [21, 22] , anti-HCV [23], hypoglycemic [24-26] and have a microtubule-stabilizing CNS [26], [34]. Many biological applications make use of 1,2,4-triazolo [1,5-a] pyrimidine derivatives[27-32]. The spectroscopic properties of triazolo pyrimidine derivatives in the ultraviolet-visible (UV-vis) region were theoretically explored using Time-Dependent Density Functional Theory (TD-DFT) techniques to illustrate the influence of solvent polarity on UV-vis spectra [26]. The spectroscopic characteristics of triazolo pyrimidine metal complexes were also computed using the DFT and TD-DFT techniques [33, 34]. The electronic structure and UV–vis spectra of ruthenium(II) complexes bound to triazolo pyrimidine ligands were characterized using TD–DFT techniques [35, 36]. The anionic and neutral molecular orbital derivatives were constructed using molecular orbital theory at its B3LYP/6-311+G (d, p) level. Furthermore, the geometrical parameters obtained by DFT were compared to those obtained by X-ray structures [37, 38] and found to be in excellent agreement. The molecular orbitals of triazolo pyrimidine derivatives in the ground and excited states were also estimated at the B3LYP/6-31G level of theory [39, 40].
- Experimental part for synthesis of triazolo pyrimidine
2.1. Synthesis of [1,2,3] triazolo[4,5-d] [1,2,4] triazolo[4,3-a] pyrimidine derivatives
3-benzyl-5-chloro-7-methyl-3H- [1,2,3] triazolo [4,5-d] pyrimidine may be produced by reacting benzylamine with boiling PrOH and then, diazotizing it in the presence of NaNO2/HCl (3). Compound 3 was treated in boiling EtOH with hydrazine hydrate to provide the 2-hydrazino-substituted derivative (4) demonstrated in Scheme 1 [41]. Chemical (4) reacts with CS2 in dry pyridine to produce benzyl-9-methyl-3H [1,2,3] triazolo [4,5-d].triazolo [4,3-a]. 1-benzyl-4-methyl-1H-[1,2,3] triazolo [4,5-e] isomeric pyrimidine-7 (6H)-thione (5) 1-benzyl-4-methyl-1H-[1,2,3] triazolo [4,5-e] Pyrimidine-7 (6H)-thione, Triazolo [1,2,4] triazolo [4,3-a] pyrimidine-8 is demonstrated in Scheme 2 [42].(7H)-thione (7).

Scheme 1. Synthesis of 2-hydrazino-substituted derivative

Scheme 2. Synthesis of benzyl-9-methyl-3H [1,2,3] triazolo [4,5-d] [1,2,4] triazolo[4,3-a] pyrimidine-7(6H)-thione
2.2. Synthesis of [1,2,4] triazolo [4,3-a] pyrimidine -5(1H) ones
In the presence of acetic acid, substituted 3,5-diamino [1,2,4] triazoles (9) react with beta keto esters, resulting in the addition of the keto ester to the more nucleophilic 3-amino group, followed by heterocyclization of enaminoester (10) to yield 2-substituted 3-amino [1,2,4] triazolo [4,3-a] pyrimidines (11) [41]. Triazolo pyrimidines undergo a more stable thermodynamic rearrangement to 1-substituted 3-amino-[1,2,4] tri-azolo [4,3-a] pyrimidines when heated (4) [42] Dimroth rearrangement can also occur, yielding 1-substituted 2-amino[1,2,4]triazolo[1,5-a]pyrimidines (15) [46]. This also happens when diamines (9) react with other 1,3-dicarbonyl compounds, as depicted in Scheme 3 [43]. By reacting 3-alkylamino-5-amino-1-phenyl [1,2,4] triazoles (6a, b) with-keto esters (15a-c) and diethyl ethoxymethylenemalonate (15d), amino [1,2,4] triazolo pyrimidines with an alkyl substituent at the nitrogen atom of the pyrimidine or at the endocyclic amino group are produced. (Triazoles (14a, b) are heated with esters (15a-d) in the presence of acetic acid to produce 3-alkylamino-1-phenyl [1,2,4] triazolo[4,3-a] pyrimidin-5-ones, as shown in Scheme 4 (16a-f). The yields of triazolo pyrimidines (16a-f) compounds are indicated in Table 1. Alkylamino triazoles (14a, b) react with dicarbonyl compounds, as shown in Scheme 5 (15a-d). We may get triazolo pyrimidine from enaminoester (17e) by heating it at reflux in ethanol and then, heating it in the presence of acetic acid (16e). Condensation of triazoles (14a, b) with esters (15a-d) yielded triazolo pyrimidines (16a-f). The synthesis of enaminoesters (17e), which then cyclize to yield triazolo pyrimidines (18) as intermediates, appears to be the first step. Triazolo pyrimidines (18) recombine to generate more stable triazolo pyrimidines (16). 2-imino [1,2,4] triazolo [1,5-a] pyrimidines can be made by an unstable alternate chemical method (19) [44].

Scheme 3. Synthesis of 2-substituted 3-amino [1,2,4] triazolo[4,3-a] pyrimidines

Scheme 4. Synthesis of 3-alkylamino-1-phenyl [1,2,4] triazolo[4,3-a] pyrimidin-5-ones
Table 1. Yields of triazolo pyrimidines (16a-f) compounds [45]


Scheme 5. Synthesis of triazolo pyrimidines (16a-f) and compounds 20-22
2.3. Synthesis of [1,2,4]-triazolo[1,5-a] pyrimidines
The oxidative cyclization of appropriate N-benzylidene-N-pyrimidin-2-yl hydrazine precursors was followed by the preparation of novel [1,2,4]-triazolo-[1,5-a] pyrimidine derivatives. As demonstrated in Scheme 6 [45], a Dimroth adjustment occurs.

Scheme 6. Synthesis of [1,2,4]-triazolo-[1,5-a] pyrimidine derivatives
We made hydrazine (38) from 2-chloro-5-bromopyrimidine (37) and condensed it with substituted benzaldehydes to get hydrazones (39). Hydrazones (39a–d) were cyclized to [1,2,4]-triazolo[4,3-a] pyrimidines (40a–d) using [1.1]. By reacting the 6-bromo[1,2,4]-triazolo[1,5-a] pyrimidines (40a–c) with alkyl amines, compounds (41a–c) were formed. The hydroxyl derivative was obtained by demethylating compound (41a) with BBr3 (41d). As indicated in Scheme 7, MW irradiation of the bromo derivative (40d) with an aliphatic amine produced the 7-amino derivatives (43e–g). The van der Plas’ ANRORC process [46-48] occurred when the substitution occurred in the t position of the leaving group.

Scheme 7. Synthesis of (43e–g) derivatives
2.4. Synthesis of [Re (CO)3L2Cl] (L = 1,2,4-triazolo-[1,5-a] pyrimidine)
(0.2 g, 0.55 mmol) of Re(CO)5Cl and (0.114 g, 1.16 mmol) of 1,2,4-triazolo-[1,5-a] pyrimidine were refluxed for 6 hours in toluene (80 ml) [49].
2.5. [1,2,4]-Triazolo[1,5-a] pyrimidine derivatives
Inhibition of mild steel corrosion was carried out by 1,2,4-triazolo[1,5-a]pyrimidine derivatives (P3 and P4) in 1 M HCl solution [50].
2.6. Synthesis of [1,2,4] triazolo-[1,5-a] pyrimidine and 5,7-dimethyl- [1,2,4] triazolo- [1,5-a] pyrimidine
[1,2,4]triazolo-[1,5,a]pyrimidine (tp) and 5,7-dimethyl-[1,2,4]triazolo-[1,5,a]pyrimidine (dmtp) react with diorganotin dichloride to form Me2SnCl2(tp)2, Et2 SnCl2(tp)2, Me2 SnCl2(dmtp)2, Et2 SnCl2(dmtp)2, Bu2SnCl2(dmtp), Ph2SnCl2(dmtp) [51].
2.7. Synthesis of triazolo[1,5-a] pyrimidine derivatives
The seven compounds were made using methods described in the literature [52]. Two fused triazolo pyrimidine rings are connected to two terminal phenyl groups through a pyrimidine ring in the suggested molecules. The effect of substitutes is explored. The structure of the seven suggested triazolo [1,5-a] pyrimidine derivatives (45–51) is revealed by the insertion of different substituents attached beside position 4 to both phenyl groups, Ph-X and Ph-Y, in which molecule. As demonstrated in Scheme 8. (45) is 5,7-diphenyl-4,7-dihydrofuran-[1,2,4]. 5-(4-fluorophenyl)-7-(4-nitrophenyl)-4,7-dihydro-[1,2,4] pyrimidine (46) is 5-(4-fluorophenyl)-7-(4-nitrophenyl)-4,7-dihydro-[1,2,4] pyrimidine (46) is 5-(4-fluorophenyl)-7-(4-nitrophenyl)(47) is 5-(4-fluorophenyl)-7-(4-methoxyphenyl)-4,7-dihydro-[1,2,4] triazolo [1,5-a] pyrimidine, (48) is 5,7-bis(4-fluorophenyl)-4,7-dihydro-[1,2,4] triazolo [1,5-a] pyrimidine, and (49) is 7-(4-brom(50) is 5-(4-fluorophenyl)-7-(p-tolyl)-4,7-dihydro-[1,2,4] pyrimidine triazolo.

Scheme 8. Synthesis of triazolo[1,5-a] pyrimidine derivatives which, under investigation in this part
2.8. Synthesis of amino [1,2,4] triazolo[1,5-a] pyrimidines
To get mesoionic compounds (57a-e) as well as (56c-f), cations of compounds (55a-e) and (56c-f) were deprotonated at the amide nitrogen in aqueous sodium carbonate solutions at ambient temperature (58c-f). As demonstrated in Scheme 9[53] , cations of compounds (59a,g,h) lost a proton from the NH group of the dihydropyrimidine moiety to provide free bases (60 a,g,h).

Scheme 9. Synthesis of free bases of (60a, g, h) compounds
2.9. Synthesis of amino [1,2,4] triazolo[1,5-a] pyrimidines
By reacting 2-aminosubstituted [1,2,4] triazolo[1,5-a] pyrimidines (61) with their counterparts in acid medium with varying pyrimidine ring saturation and 1,3-diketones or 1,1,3,3-tetramethoxypropane, variably substituted polycyclic derivatives of triazolo pyrimidine were synthesized. As demonstrated in Scheme 10. The reaction of 4,5,6,7-tetrahydro- or aromatic amino triazolo pyrimidines with a cascade rearrangement with recyclization of the dihydropyrimidine ring yields partially hydrogenated [1,2,4]triazolo[1,5-a:4,3-a']dipyrimidin-5-ium salts, and the reaction of substrates containing the4,7-dihydro-[1,2,4]triazolo. As demonstrated in Scheme 11[54].

Scheme 10. Synthesis of [1,2,4] triazolo[1,5-a:4,3-a`] dipyrimidin-5-ium or 1,2,4] triazolo[3,4-b] quinazolin-5-ium salts

Scheme 11. [1,2,4] triazolo[1,5-a:4,3-a`] dipyrimidin-5-ium or 1,2,4] triazolo[3,4-b] quinazolin-5-ium salts yield
2.10. Amino [1,2,4] triazolo[1,5-а]-pyrimidines
2-Amino-substituted triazolo pyrimidines (73–75) with various saturation of the pyrimidine fragment (Scheme 12) is used for the synthesis of substituted triazolo pyrimidines [1-3] and their polycyclic derivatives [4-7]. Compounds (73–75) were obtained by reactions of 3,5-diamino-1,2,4-triazole with many saturation of the pyrimidine ring was obtained by oxidation or hydrogenation of the dihydro derivatives as shown in Scheme 12 [55-57].

Scheme 12. Synthesis of 2-amino-substituted triazolo pyrimidines
2.11. Synthesis of R-7-methyl [1,2,4] triazolo[2,3-a] pyrimidines
5-R-3-amino-1,2,4-triazoles (76) regioselectively reacted with ethoxymethylidene acetylacetone and ethyl ethoxymethylidene acetoacetate to provide 2-R-7-methyl [1,2,4] triazolo[2,3-a] pyrimidines, as indicated in Scheme 13. Reflux of compounds (76a-f) with ethoxy methylidene acetylacetone or ethyl ethoxymethylidene acetoacetate in acetic acid for 40-60 minutes provides the corresponding 2-R-7-me-thyl[1,2,4]triazolo[2,3-a]pyrimidines (84) in high yields, regardless of the substituent R, as indicated in Scheme 14 [58].

Scheme 13. Synthesis of 2-R-7-methyl [1,2,4] triazolo[2,3-a] pyrimidines

Scheme 14. Yield of 2-R-7-methyl [1,2,4] triazolo[2,3-a] pyrimidines and 2-R-7-methyl [1,2,4] triazolo[2,3-a] pyrimidines
2.12. Synthesis of thieno[3,2-e] [1,2,4] triazolo[1,5-c] pyrimidines
Both nucleophilic and electrophilic agents can trigger the recyclizations, which follow the same steps: nucleophile (electrophile) ring opening ring closure (paths A and B) as indicated in Scheme 15 [59, 60].

Scheme 15. The recyclizations paths of the addition of nucleophile (electrophile) ring opening ring closure (paths A and B)
2.2. Computational evaluations
2.2.1. (6a–e) and (8a–e) compounds
DFT calculations in the solvent phase were performed using the Polarizable Continuum Model (PCM) at the B3LYP-6-311+g (2d, p) level of theory to compute the proton chemical shifts of (6a–e) and (8a–e) compounds. The resulting results agreed well with the experimental data. (6a–e) compounds are more thermodynamically favorable than (8a–e) compounds based on the relative Gibbs free energy [61, 62]. Table 2 indicates the chemical shifts of the two benzylic hydrogens on the carbon linked to nitrogen N-3 in (8a–e) or nitrogen N-1 in (8a–e). Cyclisation reaction occurs in such a way that the anthracene-like hetero-cycle is formed.
Table 2. Experimental and theoretical 1HNMR chemical shifts in ppm relative to TMS for benzylic protons (on carbon attached to nitrogen N-3 of (8a-e) or nitrogen N-4 of (6a-e) in PCM (chloroform)/ B3LYP/6-311+g (2d, p)

a Adjacent proton to nitrogen N-4 of 6a–e.
b Adjacent proton to sulfur atom in (8a–e).
2.2.2. Compounds (20-22)
To determine the thermodynamic likelihood of production of isomers (8), (10), and (12), quantum-chemical calculations for the energy parameters of model compounds (20), (21) and (22) were performed (11). Table 3 displays the computed relative Gibbs energies (G298), dipole moments, and relative concentrations (x) of hypothetical equilibrium mixes of isomers (20, 21, and 22) in gas and aqueous phases. It was discovered that isomer (20) is more stable in nonpolar solvents and aqueous solutions than isomers (21) and (22) and that its equilibrium concentration approaches 100%. Isomer (21) is thermodynamically more stable (3.9-5.0 kcal/mol) than isomer (22) due to intra-molecular hydrogen bonding. The difference in free energy between isomeric amino triazolo pyrimidines 3-5 (3.6-4.9 kcal/mol) and alkyl amino triazolo pyrimidine (20) and imino isomers (21) (9.8 kcal/mol) and (22) (13.6 kcal/mol) is greater than the difference in free energy between isomeric amino triazolo pyrimidines 3-5 (3.6-4.9 kcal/mol) [63].
Structures of model molecules (20), (21), and (22) (intra-molecular hydrogen bonds are indicated in the Scheme 5).
Table 3. Relative Gibbs Energies ΔG298, Dipole Moments µ and Relative Concentrations x in Equilibrium Mixtures of Isomers (20), (21) and (22) at 298K

2.2.3. [1,2,4] Triazolo[1,5-c] pyrimidine
[1,2,4]-triazolo[4,3-c] pyrimidine rearrangement to [1,2,4]-triazolo[4,3-c] pyrimidine rearrangement. The hypothesis of triazolo[1,5-c]pyrimidine was investigated at the B3LYP/6-31G(d,p) level [64]. Diverse processes (neutral, acidic, and basic), as well as solvent effects, all point to an ANRORC mechanism for the Dimroth rearrangement. The right-side isomers are more stable than the left side isomers in the three equilibriums of (Scheme 8): XI contains 48.4 kJ mol-1, XIII has 50.1 kJ mol-1, and XV contains 28 kJ mol-1. As depicted in Table 4, 43a–g the [1,5-a], isomer is more stable than [4,3-a] between 60 and 62 kJ mol-1. Table 5 reveals that the [1,5-a] isomers are more stable than the [4,3-a] isotopes between 57 and 69 kJ mol-1 [65] for 6-bromo derivatives (40a–d).
Table 4. Total energy (Hartree) and relative energy (kJ mol-1) of the [1,2,4] triazolo[1,5a] pyrimidine, (43a–g), and the (hypothetical) [1,2,4]-triazolo [4,3] pyrimidine isomers (43a–g bis)

Table 5. Total energy (hartree) and relative energy (kJ mol-1) of the [1,2,4]-triazolo [4,3- a] pyrimidine, (40a–d), and their [1,2,4]-triazolo[1,5-a] pyrimidine counterparts, (40a–d bis) [70]

2.2.4. [1,2,4]-triazolo [1,42 a] pyrimidine isomer
Table 6 indicates theoretical statistics for the hypothetical [4,3-a] isomer of (42a) (42a bis). We find that the estimated 13C and 15N chemical shifts for the [1,2,4]-triazolo [1,42 a] pyrimidine isomer geometry match those found experimentally, however the states of (5b and 5c bis) differ significantly, as reported in Tables 7, 8, 9 and 10 [66, 67].
Table 6. Measured chemical shifts of compound (5a), and absolute shielding and calculated chemical shifts for the [1,5-a] and the (hypothetical) [4,3-a] structures


Table 7. Absolute shielding, calculated and measured chemical shifts of compound (5b and 5c)


Table 8. The experimental and optimized bond lengths [Å] and angles [°] for [Re (CO)3(tp)2Cl]

Table 9. The energy and molar absorption coefficients of the experimental absorption bands and the singlet electronic transitions calculated with the TD-DFT method and basis B1 for [Re (CO)3(tp)2Cl]


Table 10. The vertical excitation energy of the optimized lowest singlet and triplet states and bond lengths between Re and ligand atoms for [Re (CO)3(tp)2Cl] in basis B2
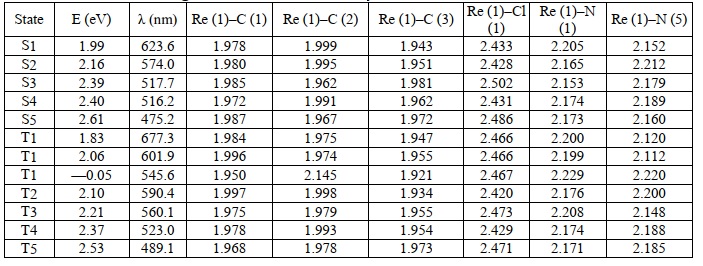
2.2.6. R2SnCl2 (tp)2 compounds (R = Me, Et)
The two lowest energy structures were created using DFT calculations on R2SnCl2(tp)2 molecules (R = Me, Et). Table 11 depicts their key geometrical characteristics, relative energy, and computed DE values. As demonstrated in Table 11, the structure of the cis-Cl2 isomer is more stable in gas than the trans-Cl2 isomer for Me2SnCl2(tp)2 and Et2SnCl2(tp)2 compounds by kJ/mol. The preferred geometry is Me2SnCl2(tp)2 _B, i.e., the all-trans geometry, and Et2SnCl2(tp)2 A, i.e., the trans-diethyl, cis-Cl2 geometry, based on a comparison of computed and observed nuclear quadrupole splitting values. In fact, there may be a preferred understanding of the structure of the compound Et2SnCl2(tp)2, as displayed in Table 12, between the experimental DE, 3.45 mm s-1, and the computed worth relative to the majority stable structure, agreement between the experimental DE, 3.45 mm s-1, and that computed to Et2SnCl2(tp)2 _B, 4. 10 mm s-1, as shown in Table 13. For the same structural environment, there is good agreement between the computed value of the C–Sn–C angle of Et2SnCl2(tp)2A, 136.4o, and the one assessed by the point charge formalism, 141o [70, 71]. In the case of Me2Sn- Cl2(tp)2, however, the desired structure would not be the most energetically stable in vacuo, i.e., Me2SnCl2(tp)2A, since Me2SnCl2(tp)2 B has a considerably better agreement between the experimental and predicted DE, i.e., 4.16- and 4.15-mm s-1, respectively (see Tables 11 and 12).
Table 11. 119Sn Mo¨ ssbauer parameters of diorganotin (IV) chlorides and their tp and dmtp adducts

“a” In the solid state and at liquid-nitrogen temperature, unless otherwise specified.
“b” Isomer shift with respect to room-temperature CaSnO3.
“c” Nuclear quadrupole splitting.
“d” Full width at half-height of the resonant peaks.
Table 12. Geometrical parameters (bond distances, in A˚, and angles, in degrees), their relative energy and calculated nuclear quadrupole splitting values calculated at DFT level (see text) for the compounds


2.2.7. Triazolo[1,5-a] pyrimidine derivatives
Optimization of triazolo[1,5-a] pyrimidine derivatives in the gas phase using DFT at the B3LYP/6-311G (df, pd) level of theory. Table 13 [72] illustrates the EHOMO, ELUMO, energy gap Eg, and dipole moment of all derivatives. Table 14 shows the actual and theoretical electronic absorption spectra of chemical (45) in Dioxane and DMF. The theoretical gas phase transitions of the different subsystems a, b, and c were also estimated and presented in Table 15 using TD-B3LYP/6-311G (d, p). Inserting a NO2 group in position X and a F atom in position Y in Ph-X and Ph-Y of compound (46) yields compound (46) (45). Table 16 shows the experimental and theoretical electronic absorption spectra of compound (46) in Dioxane and DMF, just as compound (45). By placing the F atom in Ph-Y and the OCH3 group in position X, compound (47) is formed (46). The actual and theoretical electronic absorption spectra of compound (47) in Dioxane and DMF are displayed in Table 17. Table 18 shows the experimental and theoretical electronic absorption spectra of compound (48) in Dioxane and DMF, which was made by replacing the F atom in position X of compound (46) to make compound (48). The synthesis of compound (48) is achieved by replacing the F atom at position X with a Br atom (49). The actual and theoretical electronic absorption spectra of compound (49) in Dioxane and DMF are reported in Table 19. The synthesis of compound (46), when the CH3 group is inserted in position X, is compound (46) and (50). The actual and theoretical electronic absorption spectra of compound (50) in Dioxane and DMF are shown in Table 20. The -isoelectronic compound (51) is made by replacing the F atom with the OCH3 group of compounds (47). In dioxane and DMF [73], Table 21 illustrates the experimental and theoretical electronic absorption data of compound (51).
Table 13. Theoretical calculation of total energy, EHOMO, ELUMO, Energy gap (Eg) and dipole moment of compounds (45- 51), calculated at B3LYP/6-311G (df, pd) in gas phase

a I.E., = - EHOMO
b E.A. = - ELUMO
cFor numbering system.
Table 14. The experimental and theoretical electronic absorption spectra of compound (45) in dioxane and DMF

Table15. Theoretical vertical excitations of compound (45) and its subsystems calculated at TD- B3LYP/6-311G (d, p) in gas phase

Table16. Experimental and theoretical UV spectra of compound (46), calculated at TD-B3LYP/6-311G (d, p)


Table17. Experimental and theoretical UV spectra of compound (47), calculated at TD-B3LYP/6-311G (d, p)


Table 18. Experimental and theoretical UV spectra of compound (48), calculated at TD-B3LYP/6-311G (d, p)

Table 19. Experimental and theoretical UV spectra of compound (49), calculated at TD-B3LYP/6-311G (d, p)

Table 20. Experimental and theoretical UV spectra of compound (50), calculated at TD-B3LYP/6-311G (d, p)

Table 21. Experimental and theoretical UV spectra of compound 51, calculated at TD-B3LYP/6-311G (d, p)

2.2.8. Triazolopyrimidine derivatives
Quantum and molecular mechanics calculations indicated that the Inhibition Efficiency was related to quantum chemical parameters. The experimental Inhibition Efficiency was found to be close to that of the theoretical study. In terms of inhibitory effectiveness, (Pyrimidine thione) (53) surpasses Pyrimidinones, according to the findings (IE). Inhibition efficiency for triazolopyrimidine derivatives as corrosion inhibitors for steel in acidic media has been reported as Eexp (percent) based on the approach of weight loss for compounds (52– 53). As indicated in Table 22, compound 53 has the greatest EHOMO value, which is compatible with the experimental percent inhibition efficiency data, suggesting that triazolopyrimidine thione (53) is a better inhibitor than pyrimidinones. The addition of a sulfur atom to thione boosts efficiency when compared to the oxygen counterpart. The capacity to accept electrons is indicated by the ELUMO. As a result, the lower the ELUMO value, the better the chances of the molecule receiving electrons. In Table 22 and Tables 23, ELUMO has the lowest value (53). All the compounds discussed in this section are illustrated in Scheme 16. The correlation coefficients and degree of linearity between DFT-calculated quantum chemical parameters and experimental inhibitory efficacy of the Triazolo pyrimidines studied are depicted in Table 24. The theoretical inhibitory efficiencies and quantum chemical descriptors for the Triazolopyrimidine derivatives investigated using the AM1 model are shown in Table 25 and Table 26 [74, 75]. A comparison of the inhibitory efficiency of several Triazolopyrimidine derivatives demonstrates that orbital energies (EHOMO and ELUMO), Energy band gap (ELUMO-EHOMO), Dipole moment (µ), Log P, Polarizability, Softness(S), and Hardness(ƞ) are all intricately connected to their inhibition impact. With increasing EHOMO, lowering ELUMO, reducing ELUMO-EHOMO, rising Dipole moment, raising Log P, increasing Polarizability, increasing Softness, and decreasing Hardness, the inhibitory efficacy of the Triazolopyrimidine derivatives improves. As evidenced by the experimental data, (53) (Pyrimidine thione) had a higher Inhibition Efficiency (IE) than Pyrimidinones. A high significant coefficient of determination (R2 =0.804) was determined between experimental and inhibitory efficiency.

Scheme 16. (52-54) studied compounds
Table 22. Quantum chemical parameters of triazolopyrimidine derivatives using AM1 method

Table 23. Quantum chemical parameters of triazolopyrimidine derivatives using DFT method

Table 24. Correlation coefficients, r (degree of linearity, R2) between quantum chemical parameters and experimental inhibition efficiencies of the studied triazolo pyrimidines

Table 25. Theoretical inhibition efficiencies of the studied triazolopyrimidine obtained from AM1 model
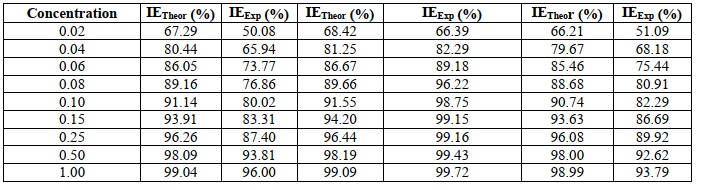
Table 26. Calculated quantum chemical descriptors for the studied triazolopyrimidine derivatives using AM1 model

2.2.9. Compounds (57a-e) and (60a, g, h)
The relative Gibbs free energies (∆G298), dipole moments (µ), and Boltzmann populations (x) of tautomer A-D compounds (57a-e) and (60 a,g,h) which calculated by the DFT method at B3LYP/6-311++G(2d,2p) in the gas, DMSO, and water phases [76]. All results obtained in this study are indicated in Table 27.
Table 27. Relative Gibbs free energies ∆G298 (kcal/mol), a dipole moments µ(D), and relative populations x (%) of tautomer A-D of model compounds (57) and (60) at 48 ○C calculated by the B3LYP/6-311++G(2d,2p) method"
 2.2.10. Compounds 66-68
2.2.10. Compounds 66-68
The Gibbs free energy (G298) for each tautomer was calculated using DFT techniques to optimize the geometry of (66-68) compounds. The 67A molecule is the most stable tautomeric, according to our findings. The following relationship was used to calculate the equilibrium compositions of mixes of isomers using conventional Gibbs free energy calculations: ΔG = −RT ln K. The values of ΔG298 and the relative concentrations of the isomers in aqueous solutions. These factors had no effect in vacuum or DMSO solutions on Molecules (67A) and (68), which are much more stable isomers in the relevant equilibrium. The experimentally observed kinetic product 16n to thermodynamic product 11n rearrangement is in good accord. In cyclo condensations, the production of isomers 66 and 67 appears to be predicted [77]. Although in the case of R = H, the model cations 67 are somewhat more stable than (66), the presence of bulky substituents in the actual molecules moves the equilibrium to the side of isomers (66), as observed in the case of R = Ph.
2.2.11. Amino [1,2,4] triazolo- [1,5-a] pyrimidines (73a,74a,75a)
Model compounds for theoretical reactivity assessment using DFT techniques were 2-amino [1,2,4] triazolo- [1,5-a] pyrimidines (73a,74a,75a). The global nucleophilicity of amino triazolo pyrimidines increases with increased pyrimidine ring saturation, according to most reactivity indices, as indicated in Table 28. The aromatic compound (75a) depicts a greater rise in nucleophilicity than the dihydro derivative (74a), whereas the nucleophilicity of compounds (73a) and (74a) was virtually identical. During frontier-controlled interactions, the Fukui functions fk¯ reflect the reactivity of molecules. The N-1 atom and amino group should be the most nucleophilic sites in compounds (73–75a) towards soft electrophiles, according to the data in Table 29, whereas the N-4 atom should be the most nucleophilic site in compounds (73a) and (74a). When a polar solvent (water) is compared to a nonpolar medium (gas), the reactivity of the N-1 atom should rise, whilst the reactivity of the amino group should decrease in compounds (73a) and (74a) but increase in compound (75a). In aqueous solution, the relative nucleophilicity of the N-4 atom in molecules (73a) and (74a) is also somewhat greater. As demonstrated in Scheme 17. Table 29 [78] lists all of the compounds examined in this section.

Scheme 17. Amino [1,2,4] triazolo- [1,5-a] pyrimidines (73a,74a,75a) studied compounds
Table 28. The global reactivity indices of compounds (73a, 74a, 75a), calculated at the DFT B3LYP/6-311++G(2d,2p) level of theory in gas phase and aqueous solution

Table 29. The Fukui functions fk¯ for electrophilic attack and local nucleophilicity values* Nuk(i) for compounds (73a,74a, 75a) calculated at the DFT B3LYP/6-311++G(2d,2p) level of theory in gas phase and aqueous solution

Local nucleophilicity values were calculated according to the equation Nuk(i) = Nu(i)·fk¯.
2.2.12. System (101) with R =CnH2n+1
2.2.12.1. A-Rearrangement in an acidic medium
As indicated in Table 30, DFT calculations were used to calculate the electron chemical potentials, chemical harnesses, and global electrophilicity for system (101) with R =CnH2n+1, where n = 0, 1, 2, 3, or 4, which are used as efficient static descriptors for the prediction of the reactivity of this system. This is consistent with the results of experiments on compound (98a) isomerization in acidic environments. The simulations also demonstrated that the interaction of (101) with such an electrophile is controlled by charges on atoms at long distances, whilst the reaction is controlled by the HOMO of the substrate at small distances [79, 80]. The contributions of the substrate’s pz-AO to HOMO and HOMO are depicted in Table 31. (LUMO). The relative energies of the seven cationic adducts, as well as the lengths of the C (5)-N (4) link broken during recyclization, are shown in Table 32. In terms of the polarizable continuum model, Table 33 estimated and summarized the solvation effects in the acid catalyzed rearrangement (PCM). Potential energy surface for proton-catalyzed rearrangements (103, 108) in the gas phase (solid line) and in solution (dashed line) or is the reaction coordinate is provided in Scheme 18, and the structures of these compounds are illustrated in Scheme 19.

Scheme 18 Global electrophilicity for system (101)
Table 30. Electron chemical potentials (µ), the chemical harnesses (ŋ), and the global electrophilicity indices (ω) for system (101) with R = H, Me, Et, Pr, and Bu

Table 31. Contributions of pz-AO to the frontier MO of system (101) with R = H, Me, Et, Pr, and Bu

Table 32. Energy and geometric characteristics of the possible adducts of (101) (R = H) with a proton

Table 33. Total energies (Etot), the relative total energies (Erel), and the Gibbs free energies (∆G) calculated for the gas phase of systems (103) (R = H), (104), (105), and (107) and the relative Gibbs free energies calculated with inclusion of the solvent effect (∆Gsolv)


Scheme 19. Potential energy surface along the minimum-energy path for the proton-catalyzed rearrangement (103), (108) in the gas phase (solid line) and in solution (dashed line); r is the reaction coordinate

Scheme 20. The proton-catalyzed rearrangement of (103), (108)
2.2.12.1. b- Rearrangement in an alkaline medium
The nucleophilic attack on the electron deficient C (5) atom positioned between two electronegative nitrogen atoms, N (4), and N (6), would be predicted to occur already in the first step in an alkaline media, as opposed to an acidic medium. As indicated in Scheme 20, this produces anion (108). Total energies (Etot), relative total energies (Erel), and Gibbs free energies (G) calculated for the gas phase of systems (103) (R = H) and (108-111) were displayed in Scheme 21 and Table 34, which displayed total energies (Etot), relative total energies (Erel), and Gibbs free energies (G) calculated for the gas phase of systems (103) (R = H), (108-111), and the relative Gibbs free energies of the solvent effect (∆Gsolv). Transition states TS1-TS3 are presented in Scheme 22. The total energy of compound (103) (R = H) and a hydroxide anion at an infinite distance from each other was taken as the reference point [79].

Scheme 21. Anion 108
Table 34. Total energies (Etot), the relative total energies (Erel), and the Gibbs free energies (∆G) calculated for the gas phase of systems (103) (R = H) and (108-111) and the relative Gibbs free energies calculated with inclusion of the solvent effect (∆Gsolv)


Scheme 22. Potential energy surface along the minimum-energy path for the hydroxide-catalyzed re-arrangement (103, 108) in the gas phase (solid line) and in solution (dashed line); r is the reaction coordinate
- Conclusion
We extract information about the structures, prosperities, preparations, characterizations, and stability of triazolopyrimidine derivatives from a collection of computer studies. The primary purpose of this research is to see if these chemicals can be used as inhibitors or in drug delivery. Calculations using quantum and molecular mechanics revealed that the inhibition efficiency was linked to quantum chemical parameters. As a result, triazolopyrimidine derivatives could be considered the lead chemicals for future medical and agrochemical development.
)