Document Type : Review Article
Authors
1 Universidad de Costa Rica, Escuela de Química, 11501-2060 San Pedro de Montes de Oca, San José, Costa Rica
2 Centro de Investigaciones en Productos Naturales (CIPRONA), 11501-2060 San Pedro de Montes de Oca, San José, Costa Rica
3 Centre for Experimental and Constructive Mathematics, Department of Mathematics, Simon Fraser University, 8888 University Drive, Burnaby, British Columbia V5A 1S6 Canada
Abstract
Hybrid atomic orbitals have become a central component of organic chemistry. The history of these hybrids and hybridization as a process is discussed; this history is divided into a timeline from before 1931 to the present day. The confusion among various descriptions of ‘hybridization’, which has perplexed generations of chemists, is clarified. Based on a critical survey of the literature, including the latest experimental results and our results, we summarize the main points of using hybridization from the past and why hybridization should be eliminated from organic chemistry. Even though hybrid orbitals might have fulfilled a role as crude models in the twentieth century, we predict that they will not survive in this new century as more complicated models are required and applied.
Graphical Abstract
)
Keywords
Main Subjects
- Introduction: Before 1930
This review of hybrid atomic orbitals and hybridization is inspired by our recent work on a critique of these hybrid orbitals [1,2]. Nearly a century has elapsed since the discovery of the process of hybridization of atomic orbitals; a critical review is essential to organize the pertinent information in the twenty-first century. This review is not impartial; as all previous reviews were biased toward support of hybridization, we contend that it is time to review all previous evidence of hybridization through a critical eye.
Hybrid atomic orbitals (HAO) are perceived to arise from a culmination of two convergent themes in scientific research, one chemical and the other physical, beginning in the nineteenth century. Half a century after Dalton’s atomic hypothesis, Archibald Scott Couper, a Scottish chemist working in Paris, proposed the first enduring notions about molecular structure: "New Chemical Theory" in 1858 [3]. Couper’s idea was that carbon atoms can link to each other following valence regularities. In 1852 Edward Frankland had already published a paper that proposed the idea of chemical valencies, hence discovering the chemical bond [4]. Within the next two decades, to explain optical activity van’t Hoff and Lebel in 1873 concurrently deduced the idea of directed valences in the form of a carbon atom having tetrahedrally oriented neighboring atoms [5]. After J. J. Thomson’s discovery of the electron in 1897, his association of an electron with the structure of an atom originated a connection between an electron and bonding, later reinforced by Rutherford’s construction of the nuclear atom [6]. G. N. Lewis associated a chemical bond with a pair of electrons between adjacent atomic nuclei [7], but Bohr recognized that a static distribution of charges must be unstable if Coulomb’s law that a force is inversely proportional to a distance between two electric charges is applicable [6].
In a brilliant intellectual achievement, Schroedinger developed wave mechanics in four papers published in 1926 [8]; with the immediately preceding matrix mechanics of Heisenberg and the symbolic method of Pauli, these articles were the foundations of quantum mechanics. In 1928, Pauling tried to relate this newly created quantum mechanics with valence. The quantum-mechanical explanation of valence is more detailed and correspondingly more powerful than the old picture. In the case of some elements of the first row, the interchange energy resulting from the formation of bonds involving shared electrons is large enough to change the ‘quantization’. “It has further been found that, as a result of the resonance phenomenon, a tetrahedral arrangement of the four bonds of the quadrivalent carbon atom is the stable one” [9].
At this point, we must recognize that quantum mechanics, as distinct from quantum physics that treats the observation and analysis of discrete phenomena, is neither a chemical theory, nor even a physical theory, but a collection of methods for calculations, or algorithms, applicable to systems on an atomic scale. At least thirteen such methods are known [10], of which at least four have been applied to the hydrogen atom (Table 1). Although some chemists might be under an illusion that quantum mechanics arose from a postulate by de Broglie [11] that a moving particle of matter has wave properties, that feature certainly inspired Schroedinger (after a hint by Debye) to develop wave mechanics, but not the preceding authors of quantum mechanics, Heisenberg and Pauli. Some formulations involve quantities that are directly subject to a multiplicative commutation property, such as matrix mechanics based on explicit matrices for coordinate and momentum, quaternionic and octonionic quantum mechanics based on the respective properties of quaternions and octonions, and wave mechanics that reflect that coordinate x and its derivative operator d/dx fail to commute: x dy/dx. ≠ d(xy)/dx.
Table 1. Mathematical methods for calculations within quantum mechanics, and their founders
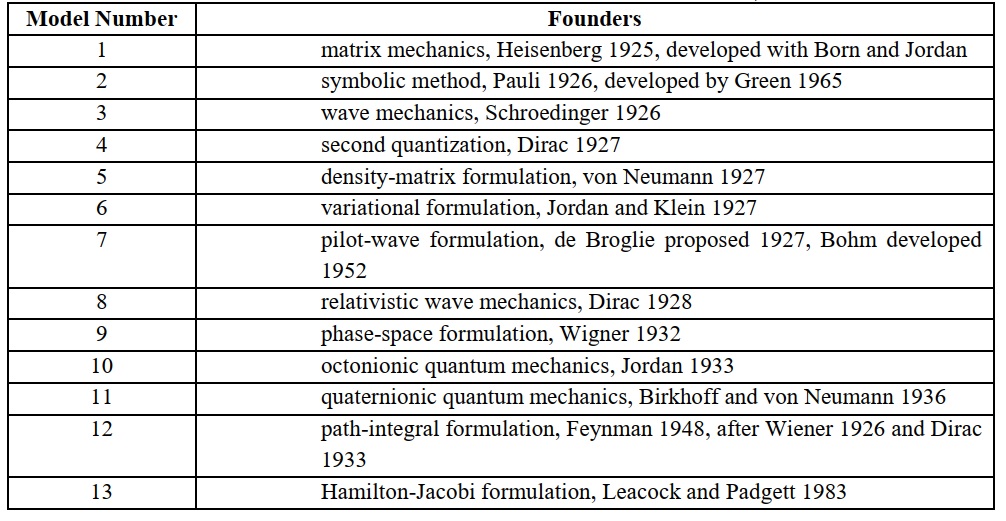
From the beginning of his acquaintance with quantum mechanics, Pauling considered that this mathematical formalism in various forms might serve as a mechanism to explain molecular structure. Although Pauling was aware of matrix mechanics, included in the last chapter of Introduction to Quantum Mechanics, for Pauling, quantum mechanics was Schroedinger’s wave mechanics [12]. Pauling associated an amplitude function, as a solution of Schroedinger’s equation independent of time, with an electron in an atom, rather than pertaining to the system as a whole; he likewise associated an electron with a particular amplitude function, even though electrons are indistinguishable.
We scrutinize orbitals and their application. In 1932 Mulliken originated the term orbital, to mean “something as much like an orbit as is possible in quantum mechanics” [13]. Apart from Mulliken’s obfuscation in this definition, it is erroneous because an orbital is an artifact not of quantum mechanics in general but of one particular method – wave mechanics – among the several methods that comprise quantum mechanics, and has no meaning or significance within the other methods. An orbital is precisely a solution of a Schroedinger equation for the hydrogen (or other one-electron) atom, and is hence neither more nor less than an algebraic formula, such as Ne−r/a˳, to which might be attached a name such as y1,0,0; such an algebraic formula has an infinite spatial extent and is intrinsically intangible. In that formula r implies the distance between an electron and an atomic nucleus, a0 indicates a physical constant known as Bohr radius and N is a normalizing factor. For Dirac’s equation in relativistic wave mechanics, each solution for the H atom as an algebraic formula, or precisely a vector with four components each of which is an algebraic formula, corresponds to a particular state of the H atom with its energy and angular momentum specified in terms of four quantum numbers, expressed concisely as | n, l, j, mj >, but, for solutions of a Schroedinger equation, no such correspondence in general exists. For both the Schroedinger and Dirac solutions in the customary spherical polar coordinates, in general the algebraic formulae are complex, i.e., having both real and imaginary parts; the latter property defies direct observation in a physical world. An association of such an orbital – an algebraic formula – with an atom or molecule as tangible matter is hence a logical error. The logical domain of an orbital is within a mathematical calculation, separate from the domain of observable quantities, such as with a microscope. In the exceptional case in which an orbital corresponds to a particular physical state of a hydrogen atom, the square of such a formula, as a product of formula y and its complex conjugate y*, has, however, a physical meaning: y* y multiplied by electronic charge e to produce e y* y dt measures the density of electronic charge in volume element dt in the vicinity of an atomic nucleus. No physical formula, such as e½ y, that contains the square root of e is known; an orbital has, therefore, no direct physical significance, reinforcing its nature as an artifact of a particular method of calculation. Any correct application of one or other method within quantum mechanics must yield the same quantitative density of electronic charge in the hydrogen atom in its particular state of defined energy and angular momentum. The orbitals as algebraic formulae that might conventionally be presumed to apply to other atoms are derived from the solution of the Schroedinger equation for H in spherical polar coordinates; these eigenfunctions, in their direct complex forms, have well defined values of orbital angular momentum and its z-component, but these angular-momentum properties are irrelevant and useless for an (improper) application of these formulae within molecules. Equally valid alternative solutions [14] of the Schroedinger equation for H in other systems of coordinate can be applied just as effectively for any descriptive purpose, even though a relation between a particular amplitude function and its angular-momentum properties might be lacking.
An extrapolation of orbitals for direct use in a qualitative description of an atom with more than one electron is deprecated at least as much as any other extrapolation. There is no objection to the use of orbitals as basis functions in explicit calculations involving other atoms –such calculations of observable properties can be valid and valuable. These incontrovertible scientific facts must serve as a foundation of chemical thinking about the electronic structure of atoms and molecules in the twenty-first century.
- Discovery: 1931
Was hybridization discovered or invented? In Pauling’s own words, “I discovered (or invented) hybridization” [15]. He subsequently stated, “quantum mechanics may be considered to have been ‘discovered’.” [16]. Considering that quantum chemists [17,18], philosophers [19] and historians [20] recently used this phrase “discovery of hybridization”, in this review we continue with this usage, but the origins of hybridization are not so simple.
Linus Pauling was a physical chemist who, following his undergraduate degree in chemical engineering in Oregon USA, applied X-ray diffraction to derive the structure of crystals of various inorganic chemical compounds in his research at California Institute of Technology, for which, with concurrent studies in mathematical physics, he was awarded a doctoral degree. In a post-doctoral stage, he was awarded a Guggenheim fellowship to study under Sommerfeld in Munich; during those two years, he passed also some months in Goettingen in the vicinity of Born, in Copenhagen in the vicinity of Bohr and in Zurich in the vicinity of Schroedinger, although there is no record of Pauling ever interacting directly with these preeminent physicists. Pauling was, however, greatly inspired by the possibilities of applying quantum mechanics to the electronic structure of atoms and molecules, particularly from his acquaintance with Heitler and London in Zurich, who had undertaken first calculation of H2 as a stable chemical species [21]. Between 1926 and 1931, Pauling evidently cogitated profoundly about methods of quantum mechanics, especially wave mechanics, because the immediate success and ease of practical applicability of the latter discouraged the development of other methods.
Both Slater (1930) and Pauling (1931) separately produced a valence-bond theory as an extension of the work of Heitler and London [21]. Both Slater and Pauling had interest in an application of wave mechanics to atomic spectra; Slater published The Theory of Complex Spectra of atoms as a paper in 1929 [22]; Pauling and Goudsmit published a book of title The Structure of Line Spectra in 1930 [23]. These same five years contained the origins of quantum chemistry, which we take to signify the eventual contemporary application of computer programs based on the Schroedinger equation to calculate various properties of molecules. These calculations originated in manual form with Lennard-Jones in 1929 [24] on diatomic molecules, and were subsequently extended by Hund [25] and Mulliken [13, 26] to polyatomic molecules in generating a molecular-orbital theory.
The concept of hybrid atomic orbitals (HAO) and the related process hybridization were introduced independently by Slater [27] and by Pauling [28], but their points of view reflected their subject affiliations. Whereas Slater presented the basis of the formulae for hybrid orbitals, with qualitative examples and insightful figures, “[Slater] laid out the procedure for solving the Schroedinger equation for molecules in general, rather than carrying out computations for actual examples” [20], Pauling presented mathematical formulae and illustrations of the “quantized” orbitals as if they were derived – even though they were not so derived – directly from wave mechanics. Few calculations were actually made with this valence-bond scheme. The reason is that the non-orthogonality between the hybrid orbitals, a feature essential for the justification of Pauling’s approach, made formulating the equations for such a calculation just too complicated and challenging; even if approximations were made, but made in a consistent fashion, any consequent calculations were nearly impossible to perform. It was thus impracticable to provide a means of connecting Pauling’s ideas to the detailed equations in an unambiguous way. Slater understood this problem of non-orthogonality, but Pauling essentially ignored its impact on the prospective feasibility of quantitative calculations [29].
Although Slater [27] and Pauling [28] had independently originated the idea of HAO, their methods contrasted, likely reflecting their physical and chemical outlooks, respectively. Slater’s approach was more mathematically based on the known mechanisms of undertaking calculations, whereas Pauling had in mind more qualitative applications to explain the gross structure of molecules. Pauling generated four tetrahedrally oriented hybrid orbitals that he applied for use with carbon and nitrogen atoms to encompass the structure of methane and related molecules [28].
- Development: 1932-1950
We regard a development of the application of HAO retrospectively, i.e., in the knowledge accumulated during successive decades until the present day. In 1932 Hultgren, with acknowledged assistance from Pauling and Podolsky, extended the coverage by including d orbitals to encompass five and six equivalent bonds; for the latter six bonds, the favored geometries are either an octahedron or a trigonal prism, but Hultgren insisted that no more than six such equivalent bond functions could be formed [30].
About the same time as Slater’s and Pauling’s origination of hybrid orbitals, Mulliken [31] developed an alternative formalism based primarily on Hund’s ideas, which he described as the “molecular point of view” involving all atomic nuclei and their associated electrons, rather than an alleged electronic decomposition into particular bonds between couples of adjacent atomic centers; this approach became described as molecular-orbital theory, in contrast with the valence-bond theory of Slater and Pauling that extended the treatment of H2 by Heitler and London. In that review, Mulliken had no hesitation in invoking electrons moving in ‘orbits’ and in distinguishing electrons that are fundamentally indistinguishable. Slater, Pauling and Mulliken all assumed electron configurations based on the hydrogen atom according to the derivation in wave mechanics in spherical polar coordinates, although there is no unique electronic configuration for atoms or molecules containing more than one electron [32], contrary to the aufbauprinzip of Bohr. At least Pauling was aware of the third seminal paper of Schroedinger in the series Quantisation as a Problem in Proper Values [8], containing the solution in paraboloidal coordinates, because he cited it, for other reasons, in Introduction to Quantum Mechanics [12]. Pauling had also attended Schroedinger’s lectures in Zurich in 1927 summer during which that solution would undoubtedly have been presented, but he never disclosed the fact of the existence of alternative solutions for the hydrogen atom. In 1930 Teller recognized that the solution of the hydrogen atom in ellipsoidal (prolate spheroidal) coordinates was infinitely preferable for a bond to hydrogen in a molecule because a second atomic nucleus was readily accommodated at the second focus of the ellipsoid [33]. Mulliken was critical of Hueckel for taking into effect only electrons of one purported type –‘p’– and ignoring an equivalent other type – ‘s’, but all these authors were perfectly content to extend the ideas of Kossel, Lewis and Langmuir that a covalent bond involves two electrons occupying overlapping orbitals for the same purpose. Although the H2 molecule, most notably in the treatment by Heitler and London [21], became the prototype for molecular bonds, in the same way that the hydrogen atom furnished atomic orbitals in spherical polar coordinates that became the basis for amplitude functions on other atoms, the density of electronic charge at the center between the two protons in H2 is only about two thirds greater than the hypothetical density due to two non-interacting hydrogen atoms at the same distance; to describe this effect as a two-electron bond must be recognized to be a gross exaggeration. In other cases, there might exist smaller electronic density at the midpoint between two nominally bonded atomic centers than for the non-interacting atoms [34].
Penney originated a shift in the use of HAO in organic chemistry (Figure 1), when, in 1934, he commented on the use of sp3 hybrids for ethane and sp2 hybrids (basically using s-p orbitals that he named the “w model”) for ethene [35]. In 1935, Penney extended his calculations to ethyne and presented evidence for sp (equal proportions of s and p) hybridization to form stable bonds; for the methyl radical, he predicted a trigonal planar structure with sp2 hybridization [36]. These contributions had an enormous effect on the use of the hybridization model in organic chemistry. “Penney compared the tetrahedral model and the trigonal model by means of a valence-bond calculation and concluded that the trigonal model was energetically preferred. Although the sp3 hybrid orbitals of the tetrahedral model have greater extension along the orbital axis than the sp2 orbitals of the trigonal model, the sp3 orbitals are necessarily canted outward from the internuclear C-C line and hence overlap poorly. He subsequently proposed trigonally hybridized carbon as the building block of the s framework of the benzene ring. One may speculate that it also was based on the simple conceptual continuity of the hybridization picture (sp3 for methane, sp2 for ethene, sp for ethyne), which made the idea pedagogically attractive” [37].

Figure 1. Structures of simple organic molecules
In 1935 Van Vleck and Sherman published a review [38] that was designed to compare critically the various explanations of chemical binding. Mulliken [31] and Van Vleck [39–41] introduced the term ‘hybrid atomic orbitals’ and the related process ‘hybridization’. These authors recognized an assumption that building a complete wave function for a molecule with atomic orbitals, i.e. those calculated for the hydrogen atom in spherical polar coordinates, is an effective approximation only if the potential has strictly spherical symmetry. As an atomic wave function cannot be factored into one-electron functions, the Hartree self-consistent field, which represents the best attempt at such factorization, is not centrosymmetric if an atom has ingredients other than closed shells and s electrons. In any chemical combination there is always a distortion from central symmetry due to the fields from adjacent atoms. Like their predecessors, Van Vleck and Sherman neglected to consider orbitals in other than spherical polar coordinates, despite their prospective relevance. These factors are unimportant in a direct implementation of calculations; for instance, if orbitals serve at all as basis functions, only real functions are admitted for this purpose, despite the fact that for energy (or principal) quantum number n the complex functions numbering n2 − n outnumber the real functions for n > 2; even then they are almost invariably replaced in practical calculations by sums of Gaussian functions, whereas other calculations with density functionals can completely exclude orbitals as basis functions [42]. For qualitative explanations, the system of coordinates might be crucial. For instance, if one assumes paraboloidal coordinates, the natural orbitals of the hydrogen atom extrapolated to carbon yield exactly the same functions as the sp hybrid atomic orbitals in spherical polar coordinates – no hybridization is necessary. If one disregards, for the purpose of forming hybrid atomic orbitals, the difference in energy between the 2s and 2p levels of carbon – which is decreasingly negligible for C, N, O, any linear combination of the 2s and 2p wave functions can serve as a legitimate solution of the wave equation for the central atom. In a truly central field, mixing s and p wave functions would be an error, but, if the separation between the 2s and 2p levels is small compared with the binding energy, the ability to form the best possible bonds is an important consideration. Van Vleck and Sherman were aware that that difference of energy was not small, but is in fact larger than the energy of a C-H bond in CH4 for instance [39–41].
All these authors who were involved in forming these theories of electronic structure lacked access to modern thought that orbitals are intangible algebraic formulae, which hence belong to the category of mathematical functions, as distinct from the observable properties of molecular structure such as the lengths of putative chemical bonds and the angles between those bonds on the same atomic center. Such orbitals are legitimate components in calculations of molecular structure, but molecular mechanics can yield a comparable accuracy, for the same duration of computation, with no such artifacts [43].
Kimball [44] subsequently applied group theory to delineate the possible geometric arrangements for coordination around a central atom; he stated, for instance, that with two coordinating groups a collinear nuclear conformation was possible for sp and dp hybrid orbitals; with sd hybrids an angular conformation was necessary. Kimball included seven- and eight-coordinating atoms in his treatment, but he noted that his group-theoretical approach was unable to indicate the relative strengths of the bonds according to separate hybrid schemes [44]. He used the term “directed valence” to indicate the use of hybridized atomic orbitals that form simple geometric figures. In less than a decade, the scientific community was thus exposed from ‘quantization’ to ‘hybridization’ to ‘directed valence’, all of which had effectively the same meaning.
Walsh was among the first authors to review the correlations between hybridization, bond length and bond strength, for both C–H and C–X bonds [45]. Voge also used hybridization to rationalize the C–H bond strengths in methane [46]. Maccoll extended the relation between bond strength and hybridization and explored the observation that the order of bond strength of hybrid bonds was sp > sp2 > sp3, opposite of Pauling’s prediction [47]. We hence see how rapidly the initial presentation of hybridization spread. Twenty years after its publication, the spark of hybridization led to an explosion in almost all chemistry.
- Incorporation into Textbooks: 1949 onward
One criterion, perhaps the most important, to indicate a scientific revolution is the incorporation of a new idea in textbooks: “significant and explicit changes in the content, vocabulary and organization of textbooks before and after the period in question.” [48]. Based on that criterion, the revolution of the hybridization model spread into textbooks for organic chemistry about 1949 and continues until the present day.
As background, the publication of Pauling’s classic monograph of title The Nature of the Chemical Bond was the first foray of HAO into advanced texts for physical chemistry [49]. First published in 1939, the second edition was published between 1940-1952, succeeded by the third edition from 1960 to 1964. It is notable that, in the later editions that we examined, Pauling insisted on the formation of only sp3 hybrids for carbon; these hybrids provide the bonds for both saturated and unsaturated systems [50]. This selection required the use of tau (𝜏) bonds for multiple bonds (Figure 2). We prefer the term “tau bonds”, used in the textbook of Roberts and Caserio [51], rather than the alternative terms curved bond, equivalent bond, bent bond, banana bond or omega (Ω) bond [52].

Figure 2. Examples of tau bonds for multiple bonds
This tau model was not the standard model at the time of publication of the third edition. “Pauling put forth this conception of the structure of the double bond early in his career and stuck by it throughout his lifetime, even after the σ-π bond became the standard model. But he did not defend it in print until late in his career” [50]. For example, in the third edition of The Nature of the Chemical Bond, we read, “The greater separation of the electrons for the bent-bond structure with concentrated bond orbitals than for the σ-π structure may stabilize the bent-bond structure enough to make it the better approximation to use in discussing multiple bonds in general” [49]. He continued, however, to use outdated facts to support this position. “The picture of the carbon-carbon double bond as involving the sharing of an edge by two regular tetrahedra leads to the tetrahedral value 125°16' for the single-bond: double-bond angle. The value for this angle in both isobutene and tetramethylethylene is 123°20'.” This argument is similar to what Pauling used in his 1931 paper, using graphite as a model system: “These three bonds should lie in a plane, with angles 109°28' and 125°16'.” [28]. Pauling claimed that these preliminary experimental bond angles confirmed “the significance of the concept of the carbon atom as a regular tetrahedron” [49]. In 1959 [53] and again in 1965 [54], Bartell had experimentally measured in ethene the C-C-H bond angles (121.4°±0.6°) and the H-C-H bond angle (117.2°±0.6°), which are in stark contrast to angles 125.3° and 109.5°, respectively, predicted by Pauling’s tau model for a double bond. The tau orbital model could not rationalize the bond angles known at that time. It should be mentioned that Bartell contemplated non-bonding interactions, without hybridization, to rationalize the experimental data; these data led Bartell to declare, “Hybridization is a fraud” [55].
For this article, we reviewed textbooks in a series dedicated to introductory chemistry at the university level, with an objective to record the revolution of hybridization. It is documented that “before 1957, general chemistry textbook indices rarely contained the entries ‘Schrödinger’, ‘orbital’, or ‘hybridization’ Notable exceptions to my characterization of pre-Sputnik general chemistry textbooks are those by Linus Pauling, variously titled General Chemistry and College Chemistry and published by Freeman and Company with editions in 1947, 1950, 1953, 1955, 1964 and 1970.” [56]. For comparison, we examined textbooks of organic chemistry in only the English language available to us in the library. Several organic textbooks about the midpoint of the century included no use of ‘orbitals’ or ‘hybridization’ whatsoever: Conant and Blatt’s Fundamentals of Organic Chemistry [57], Whitmore’s Organic Chemistry [58], Wibalt’s Organic Chemistry [59], and Lowther’s Organic Chemistry: An Introductory Course [60]. During this period, extremely popular textbooks of advanced organic chemistry did not deem hybrid atomic orbitals and their assorted accoutrements sufficiently necessary to include this language for advanced students [61, 62]. It would be interesting to examine a more extensive sample of textbooks in other languages to determine a time line of the progression of hybridization.
The first instance in old textbooks of organic chemistry of the use of quantum mechanics, orbitals and hybridization that we could find was in the seventh edition of the 1949 book by Branch and Calvin [63]. In this book (of which the first edition was published in 1941), the principles of quantum mechanics were explicitly applied to organic chemistry, beginning with a few pages on the quantum-mechanical description of an atom and then providing a chapter on the quantum-mechanical description of molecular structures. This description related sp3 hybridization to the tetrahedral arrangement of the four-covalent carbon atom, sp2 hybridization to the planar structures with bond angles 120°, and sp to linear structures, with no image of the hybridized orbitals.
Another textbook from this era incorporating orbital ideas in organic chemistry was by Finar, first published in 1951; the reviewed reprinted edition was from 1963 [64]. The second chapter of this book delved into the quantum atom and the model of hybridization. “It is possible, however, to hybridise these four ‘pure’ A.O. in ways to give four valencies which may, or may not, be equivalent. Three methods of hybridization are important: i) tetrahedral (sp3 bond), ii) trigonal (sp2 bond), iii) digonal (sp bond).” [64]. Instead of limiting this description of bonding in organic compounds to Penney’s orbitals, Finar discussed also the use of Pauling’s use of tau bonds. “In ethylene, we have used sp2 trigonal hybridization (one s and one p bond) to describe the double bond. It is possible, however, to use sp3 hybridization to describe ethylene. In this case, two electrons are in one orbital of ‘banana’ shape (‘bent’ bond), with two other electrons in a second ‘banana’ orbital, equivalent to the first but the mirror image of it…In the same way, the triple bond in acetylene (previously described in terms of one s and two p bonds) may also be regarded as made up of three equivalent sp3 hybrids symmetrically disposed round the C–C axis” [64]. This book includes many black and white sketches to indicate the shape and form of the hybridized orbitals.
We suggest that the influential textbook of title Basic Principles of Organic Chemistry by Roberts & Caserio [51] swayed instructors of introductory organic chemistry to include hybridization in their classes. The first edition, published in 1964, and the second edition (published in 1977), proved to be a great success. “Chapters 5 and 9, which deal with modern structural theory, are not yet commonplace in organic texts.” [65]. The authors presented sp3 hybridization, with black and white images of the orbitals, for methane, ethane, water, methanol and ammonia. For ethene, they presented both the σ-π and the tau possibilities, but for ethyne only the σ-π model. More surprisingly, in a later textbook Organic Chemistry by the same authors [66], they minimized the hybridization information and focused on only the σ-π model: “Should we regard the two bonds as equivalent with both being bent or should we imagine that one of them – the s (sigma) bond – occupies the prime space along the bond axis and the other – the p (pi) bond – the space above and below the plane defined by the other bonds to the double-bonded carbons? Most theoretical treatments of conjugated systems make use of clouds of p electrons above and below the s bond…We shall make use of this approach” [66].
In another textbook from this era [67], Gerig stated that only carbon undergoes hybridization. He adopted only the σ-π model for multiple bonds. Furthermore, he presciently warned about hybridization: “It should be recognized that, for our purposes, the idea of hybridization is merely a very convenient device for systematizing our knowledge of the structure of carbon-containing compounds and that it is not necessarily rooted in reality” [67].
It is thus clear that, by the early 1970s, hybridization had gained entrance into introductory and advanced organic chemistry. Every succeeding textbook in organic chemistry has adopted this standard (we found no exception), typically in the first chapter. This incorporation of hybridization in the introductory textbooks of that era had not gone unnoticed: “Attempts to simplify [hybridization] result in the presentation in elementary textbooks of specious arguments, that are at best misleading and at worst incorrect. In these, the author attempts to persuade, or perhaps a better word would be to hoodwink, students into thinking that they understand a number of quite difficult and subtle ideas” [68].
Another key point is that, although early textbooks incorporated both σ-π and tau models, subsequent textbooks presented exclusively the former model. There have been attempts to re-establish the tau model in several academic papers [69, 70], but every recent introductory organic textbook we found treated the σ-π model exclusively for multiple bonds.
As an example of a textbook in advanced organic chemistry, March’s Advanced Organic Chemistry: Reactions, Mechanisms and Structures has proceeded through eight editions from 1968 to 2020. Throughout these editions, hybridization is presented in the first chapter, with the tau model relegated to a footnote. “However, most of the literature of organic chemistry is written in terms of the s–p picture, and we will use it in this book” [71].
Coulson introduced the concepts ‘hybridization ratio’ and ‘non-equivalent hybrids’ (we prefer the terms ‘hybridization index’ and ‘non-integral hybrids’) to treat cases of s-p mixing beyond sp, sp2, sp3 [72]; he later expounded on these possibilities in his book of title Valence [73]. The most extensive coverage of this topic in recent textbooks of advanced organic chemistry was perhaps given in Mislow’s Introduction to Stereochemistry, which was influential over almost 40 years (first edition 1965, reprinted sixth edition 2002). In this presentation, organic molecules such as methane, cyclopropane, ethene, ethanoyl chloride, ethyne, allene, cumulene, methanal and benzene are discussed in terms of non-integral hybrids. No reason to use this system was presented; it was a calculation a posteriori with no relation to stereochemistry: “the hybridization index is therefore determined by reference to physical properties which depend on the molecular parameters of bond length, bond angle, bond energy, bond-stretching force constant and bond moment.” [74].
The textbook by Lowry and Richardson Mechanism and Theory in Organic Chemistry [75] indicated m as hybridization index (i.e., for sp3 m=3) and states, “The angle between two hybrids completely determines the hybridization index, and conversely.” The authors related bond angle θ to the index with a trigonometric formula to calculate the fractional contribution of the atomic orbitals (Figure 3).

Figure 3. Graph of bond angle versus non-integral hybridization index to calculate spm
The advanced textbook of title Physical Organic Chemistry by Anslyn and Dougherty [76] continued in the twenty-first century to present a similar non-integral system. It is unclear whether an advantage of this system removes the original definition of mixing of fixed integral s and p orbitals and replaces it with a system of non-integral orbitals. This non-integral system using hybridization indices has been used to predict bond angles [77]; more recent evidence shows that it does not work: “The derivation of simple bond angle/sp ratio formulae is shown by calculation to be incorrect” [78].
In a succinct summary after a section on hybridization, the advanced textbook of Carey and Sundberg provided a clear caveat: “It is important to remember that hybridization is a description of the observed molecular geometry and electron density. Hybridization does not cause a molecule to have a particular shape.” [79].
- Mid-century: 1950-1981
As textbooks began to include hybridization, some research articles concurrently initiated a trend to question the use of hybridization. In this section, to save space, we review only the articles that are critical of the hybridization model.
The use of hybridization to ‘cause’ the valence of a first-row element to direct its bonds towards the attached ligands was questioned by Zimmerman and Rysselberghe [80] and by Linnett and Poe [81]. As summarized by Wu, “They show that the squares of the wave functions of the electrons of the free atoms, when put in the determinant form, are maximum when one electron is along with one of the directions of the valence of the atoms, and that when the idea of maximum overlap (exchange integral) is used, these wave functions will give the correct directed valence in the formation of molecules. The implication is that one can account for the directional property of valence without having to introduce the concept of hybridization or its equivalents.” [82].
Gray and Pritchard presented an electrostatic model instead of hybridization [83]. The thesis of Gray contains the important conclusions “in no sense can [hybridization] be said to explain molecular bond angles or dipole moments…The most puzzling feature about contemporary thought is the unquestioning acceptance of s,p,d etc., orbitals which appear from the solution of the wave equation for the hydrogen atom in spherical polar coordinates, when the problem can be just as easily solved in parabolic coordinates.” [84]. The fact that the thesis of Gray was censored and unpublished for many years was revealed only much later [85]. Boeyens subsequently commented about the entire situation, “The tragedy is that the chemistry community did not have the vision to allow such important issues to be openly debated at the time. Half a century and three generations later, it has become all that harder to re-open the discussion [about hybridization]– but be re-opened it must.” [86].
In 1957, a review of the use of hybridization concluded: “Consequently methane, which can be pictured as formed from C4- by attaching four protons, will at equilibrium be a regular tetrahedron. The electron distribution will be most conveniently described in terms of tetrahedral, sp3, hybrid orbitals, but the means of description (hybrid orbitals) should not be regarded as the cause of the molecule being tetrahedral.” [87].
In 1961, Bent reviewed the evidence of hybridization and the effect of atom hybridization on the following molecular properties: bond angles, bond lengths in nonaromatic systems, bond lengths in aromatic systems, acid strength, base strength, inductive constants, dipole moments, bond polarity, bond-stretching force constants, bond-bending force constants, bond length of carbon-oxygen double bonds, quadrupole coupling constants of chlorine and proton-13C coupling constants. He continued by stating the famous ‘Bent’s rule’: “Atomic s character concentrates in orbitals directed toward electropositive substituents” [88]. This rule is based on two errors in first assigning a hybridization to a bond, and second allowing ‘rehybridization’ of an atom according to substituents. Bent rethought supporting his own rule; in his advanced years, he wrote “The last references (those by this author) describe a non-mathematical route through chemistry to a unified model of bonding in covalent, ionic and metallic substances. Not needed is hybridization, s or p orbitals” [89].
Bartell was one of the strongest challengers of the hybridization model. In 1962, he published his own experimental and calculational observations against the use of hybridization and in favor of his own non-bonded model. “Equilibrium bond angles are poor gauges of hybridization the non-bonded model may be substituted in part, or in total, for the ‘hybridization’ model with little change in effective result.” [90].
Cook and Fowler extended Schroedinger’s historic calculations on the hydrogen atom to prolate spheroidal and spheroconal coordinates; they indicated that the latter coordinate system might be a better pedigree than the complex HAO based on spherical polar coordinates. They lamented the inconsistency of using HAO: “The central weakness of hybridization is that it is imposed on an isolated atom in an arbitrary way – the linear transformations are not seen to emerge in any natural way from the physics of the atom in a molecular environment. There is, for example, no smooth transition from the separate-atom AOs - to the molecular environment hybrids. Further, the symmetry-based rules for the formation of hybrid orbitals often generate ‘directional hybrid orbitals’ with higher symmetry than the molecule. For example, sp2 and sd2 hybrids are used for planar triangular configurations: in fact sd2 hybrids have hexagonal symmetry.” [91].
Since the discovery of hybridization, there has been almost uncritical support of this model. One example from 1986 begins with the hyperbolic introduction: “The concept of hybridization of atomic-orbital basis functions to produce spatially directed wave functions with the orientation necessary for bond formation is fundamental to the modern understanding of the molecular and electronic structure of molecules” [92]. We have shown that, during the mid-twentieth century, some dissenting arguments against hybridization appeared. The following sections expose the growth of this debate.
- The Computer Age: 1963 onward
Electronic computers in the latter half of the twentieth century created an opportunity to investigate further the calculational aspects of hybridization [93]. The questions to be asked with quantum chemistry follow. Is it possible to provide calculational support of HAO in simple molecules, or is this a cyclic argument? Is there a quantum-chemical difference between the s-p model and the tau model? Can HAO be part of the molecular-orbital framework [94]? It should be clarified that these calculations gave only approximate answers, in contrast to the results provided with the Schroedinger equation for the hydrogen atom, but were primarily based on Hartree-Fock basis sets and various localization schemes. The presentation of these calculations might have conflated the idea that ‘hybrid’ orbitals are ‘localized’ orbitals in many a chemist’s mind, as hybridization in this view is a response of an atomic core to a field of neighboring atomic cores. Because of the limitations of the computers during this period, only small molecules (e.g. H2O, CH4) were investigated.
In 1963, Peters used a crude basis set of a linear combination of atomic orbitals (LCAO-MO) to calculate localized orbitals in small molecules [95]. He stated that “the hybrid atomic orbitals (HAO) which a given atom uses in forming its two-electron bonds and lone pairs are often not orthogonal. This point is particularly striking since it has always been assumed that the HAO which an atom uses are indeed precisely orthogonal to each other. There is no theoretical or numerical justification for this assumption.” In this case, a calculation on ethane (archetypically taken as 25 % s and 75 % p character) provided a 13 % s character. Ethene (nominally 33.3 % s and 66.7 % p character) was calculated with 25 % s character and ethyne (50 % s and 50 % p character) with exactly 50 % s character: “The hybridizations in methane through benzene are remarkable for their general agreement with the conventional idea of HAO…The hybridizations reported do differ from the conventional ones in that they show larger amounts of 2p character in the HAO” [95].
Because Peters performed crude calculations, the results are not considered reliable. The main problem was that the choice of a localization condition was not unique in many cases. Other calculations in general led to varied localized orbitals; the results of Peters for H2O show this discrepancy [95]. The most robust rebuttal of his work is shown in the discussion section of an article by Jaffé: “I believe it is agreed today [1963] that the original Pauling concepts of pure sp3, sp2 and sp hybridization are only very crude approximations, with the possible exception of the most highly symmetric compounds, such as methane. In my opinion, the best way to define hybridization – to the extent that it has any real significance…is to produce equivalent orbitals.” [95].
In 1969 Trindle and Sinanoğlu were the first to attempt the characterization of s and p atomic-orbital mixing in a semi-empirical sense [96]. They applied two methods to characterize numerically the hybridization – orbital localization, and an extension of Wiberg’s bond index using molecular orbitals. The two methods gave virtually identical results in some cases: “if the set of molecular orbitals is highly localized, the bond index estimate of Xp reduces to the expression used in the direct evaluation of hybridization in a local orbital. If more than one valence-bond structure is necessary to account for all charges in the molecule, hybridization is not definable for the parts of the molecule where the structures differ” [96]. In this paper of Trindle and Sinanoğlu, the basis set of molecular orbitals was formed in calculations with complete neglect of differential overlap (CNDO-II) for several small organic molecules. For example, the local orbital analysis of methane was calculated to have 74.5±0.5 % p contribution to C-H, in striking resemblance to the 75.0 % p contribution provided by pure sp3 hybrids [96]. In contrast, the CNDO calculations for ethene provided 62.3±0.2 % p contribution to C=C, slightly less than the predicted 66.7 % from sp2 hybrids, which they ascribed to confirmation of the effect of Bent’s rule. They further provided an ‘experimental’ correlation between the calculated hybridization and 13C-H coupling parameters in NMR spectra: “Figure 1 illustrates the comparison for CH bonds in various molecules, indicating a substantial agreement among the three sets of values” [96]. Based on these results, they strongly supported the use of HAO: “The qualitative and quantitative reliability of these MO predictions of hybridization allows the conclusion that the idea of hybridization is useful outside the context of a valence-bond description of the molecular charge distribution” [96].
In 1970, Polák continued this analysis for methane, ethyne, ethene, ethane, propene, butadiene, ammonia and hydrogen cyanide. “The results of the described localization procedure applied to one-determinant closed-shell wave functions, obtained by means of ab initio SCF [self-consistent field] calculations and of the extended Hückel theory [EHT]…Both SCF and EHT treatments use a minimum basis set consisting of Slater-type atomic orbitals” [97]. Methane was calculated to have exactly 75.0 % p character for C-H bonds, ethane 73.3 %-74.2 % p character for C-C, ethene 55.8 %-63.3 % p character for C=C and ethyne 33.8 %-48.2% p character for C≡C. He concluded: “The relative difficulties in the choice of an adequate parametrization scheme may be the reason for the discrepancies in the hybrid predictions derived from SCF and EHT wave functions” [97].
In 1970 Newton et al. [98] obtained truncated localized molecular orbitals for methane, ethane, ethene and ethyne. “Most of the ab initio SCF wavefunctions employed in the current study have been presented elsewhere and are based on a minimal set of Slater-type orbitals (STO)… localization (Edmiston-Ruedenberg = ER) can be considered in three different, but equivalent, ways: maximization of intra-orbital or self-repulsion energy, minimization of inter-orbital repulsion energy, or minimization of exchange energy. We emphasize that for convenience we have chosen to discuss the intra-orbital repulsion-energy surface” [98]. For saturated methane, sp2.65 hybrids on carbon were calculated, but “The one significant exception is the carbon hybrid (sp1.93) in the ethane C-C bond” [98]; no explanation is given for the deviation of this value from sp3. Unsaturated molecules were calculated with a s-p constraint: ethene was calculated with sp1.74 C=C, and ethyne sp1.01 for C≡C. These ‘hybrid’ types reinforced the idea of Pauling-Slater hybrids.
Methane was examined to ascertain whether there could be other configurations of energy less than of the sp3 hybrid configuration. Using a Hartree-Fock program in 1971, Howat and Webster [99] determined that “the most favourable description for the carbon atom in methane when neutral is that in which it is energetically equivalent to the free atom configuration C(sp3) 5S and denoted as V4 (te1te1te1te1).”
In 1972 the authors of two papers compared methane in the Hartree-Fock localized model. Peters [100] re-analyzed methane: “We consider a 2n-electron, closed-shell molecule with n doubly occupied MO which are taken in real form throughout. Then we follow Coulson, Lennard-Jones and others by replacing the Slater determinant Ψ' of delocalized MO (ϕ') by the determinant Ψ of localized MO (ϕ)” [100]. The results of Peters were astonishing. “The carbon hybrid atomic orbital of methane is a hybrid which contains 33 % 2s character or sl/3 p2/3 or sp2…it does seem that the traditional value of sp3 for these carbon valence orbitals is an overestimate of the amount of promotion of the carbon atom…it seems that hybridization is sensitive to small changes in the forms of the basic atomic orbitals” [100]. Incongruously, the paper summarizes the work: “The results show that in many ways the two carbon-hydrogen bonds are indeed very alike and not far removed from Pauling’s description of them” [100].
Also in 1972, Clementi and Popkie [101] approached the problem of calculating the structure of methane with varied C-H bond lengths “by increasing (from zero) the R(C-H) distance, one goes from a ls2 2s2 2p6 distribution to a 1s2 2s2 2p2 2ppseudo4 distribution, and finally to a (ls2 2s2 2p2) ls4 distribution, where the set of ls4 electrons are then around the four protons (hydrogen atoms)” [101]. With “the basis set used in these computations is a Gaussian-type set,” they calculated “with charge-transfer effects, the hybridization is about s1.5p2.5.” This result is an approximate hybridization of methane as sp1.7, far from the proposed sp3 hybridization predicted from first principles. An additional calculation by Clementi and Popkie also in 1972 [102] provided hybrids s1.30p0.77 for ethyne, s1.38p1.68 for ethane, and s1.38p2.58 for staggered ethane. They commented: “The discrepancy from the ‘ideal’ ratio is very substantial; however, it is not surprising, since it has become more and more apparent in the last decade that the ‘ideal’ ratios are more of linguistic than of physical value” [102].
A change of focus from maximum strength of the hybrid orbitals (“Pauling assumed that the hybrid amplitude, i.e., the maximal magnitude of the angular part of the hybrid orbital, is a measure of the strength of a hybrid” [103]) to the maximum overlap was introduced during this period. In this method of maximum overlap, reviewed by Randić and Maksić [103], “We can thus speak of the fraction m/(m + n) of s character, and a fraction n/(n + m) of p character. Usually, however, we choose m = 1 and write hybrids as spn. In the method of maximum overlap, we search for the optimal parameters ai, bi for all hybrids of all atoms in a molecule which will maximize the sum over all bonds of suitably scaled bond overlaps…The hybrids for various hydrocarbons discussed here are calculated using Clementi orbitals and assuming either a set of standard bond lengths or using the experimental values” [103]. Cyclohexane was thus calculated to have a C-H bond sp2.87, ethene with a C-H bond sp2.17 and ethyne sp1.29. This focus gave rise to correlations between the percentage s character and the wavenumbers of vibrational stretching modes in infrared spectra, proton chemical shifts in NMR spectra, spin-spin coupling parameters in NMR spectra, proton acidities, bond energies, and bond lengths [103].
In 1972 [104], Ha claimed to show a partial C-F double-bond character in fluorinated methane by evaluating the p orbital percentage on carbon, relative to methane. “The approximate Hartree-Fock atomic orbitals employed as a basis set for the LCAO-MO-SCF wavefunctions were the Gaussian lobe function representation obtained by Whitten…relative ratio of s to p character changes successively from the sp3 to sp2 hybridization of carbon, and hence the double-bond character of the C-F bond is successively increased” [104].
In a continuation of calculations on hydrocarbons, Randić and Maksić [105] compared four semi-empirical methods with the idea that the hybrid results could be used in a future calculation ab initio. “One starts with assumed initial hybrid compositions, e.g., sp3 or sp2 or sp hybrids, and then by a systematic variation of all independent parameters approaches the optimal values… we can say that the concept of non-integer spn hybrids offers a very simple and useful model describing covalent bonding…The hybrids obtained by the MOA method might provide a good initial guess for wave functions for ab initio SCF calculations employing hybrid basis sets” [105]. In the latter work, there was no great difference among the methods: the C-H hybrid for ethane ranged from 24.6-29.3 %, ethene from 30.8-34.5 %, and ethyne from 42.2-45.0 % s character [105]. “…the results of the four different and independent approaches are so similar when the s characters of the hybrids are considered in spite of the diversity of approximations employed. The reported hybrids show deviations from the idealized canonical cases: sp, sp2 and sp3. The deviations are, however, not excessive when the bonds of less strained molecules are examined” [105]. The comparison to ‘experimental’ correlation weakened the argument; “the ΨCH hybrids describing CH bonds of the strained parts of the molecules also have the increased s characters compatible with several experimental observations…Among the experimental quantities, we selected the J(13C-H) spin-spin coupling constant, which is widely accepted as a measure of the s characters of the corresponding hybrids” [105].
Jarvie et al. [106] investigated whether hybridization was significant for the tetrahedral configuration of methane using linear-combination-of-atomic-orbitals molecular-orbital self-consistent-fie1d (LCAO-MO-SCF) wavefunctions. “To carry out the calculations, a standard LCAO MO SCF program was modified so as to keep certain specified occupied MO ‘frozen’ during the iterative process, while the remaining occupied MO were allowed to converge to optimum forms in the fixed field of the nuclear framework and the frozen MO” [106]. Their results indicated that hybridization is not important. “Therefore, we conclude that 2s → 2p promotion and/or hybridization does not cause the tetrahedral structure of CH4…In summary, we believe we have made a beginning toward a quantitative understanding of the contribution of electron promotion and hybridization to molecular conformations” [106].
Cook in 1978 created ‘General Hybridized Orbitals’ (GHO) as a choice for a basis set for unrestricted Hartree-Fock (UHF) calculations as a theory of valence; they were favorably compared to Slater-type orbitals (STO) but with limitations. “Most important, they can not (except in the ease of highly symmetrical molecules) be generated from a single set of (e.g., ns, np nd) AOs. Thus, in general, they are not orthogonal − each GHO will have a small non-zero overlap integral with other GHOs on the same centre. Also, a point to which we return later, they can be optimised separately and can be optimised separately, so the choice of an optimum GHO basis is a substantive issue even in the single-determinant MO method” [107].
Closely related to the work of McWeeny and Del Re [108] to calculate hybrids a priori in a molecule, in 1980, Weinhold used simple INDO-SCF-MO wave functions to create ‘natural hybrid orbitals’ (NHO) [109]. “In summary, our procedure consists of the following steps: (i) find the density matrix P in a basis set of atomic orbitals and diagonalize each atomic subblock PAA to find the lone-pair hybrid(s) on that centre; (ii) for each pair of atoms, A, L, form the two-centre density matrix PAL and the associated matrix P(AL) ‘depleted’ of any lone-pair eigenvectors [each doubly occupied (n > nmin) eigenvector of this matrix is decomposed to give additional directed hybrids on centre A]; (iii) symmetrically orthogonalize the hybrids found in steps i and ii to find the final natural hybrids.” These ‘natural hybrids’ gave results similar to maximum-occupancy hybrids. Ethane thus gave a C-C hybrid of 72.6 % p character, ethene 59.2 % p character and ethyne 44.8 % p character. However, as they pointed out, “NHO change continuously with the molecular environment; they are not generally transferable from one molecule to another.” This procedure seemed to fail where resonance is present (e.g., CO2, methanoic acid, benzene).
The utilization of sp hybridization was reconsidered in two papers by Magnusson in 1984 [110, 111]; calculations ab initio using MO at the single-configuration restricted-Hartree-Fock level, including basis set 3-21G, were used for most compounds in that work. In one example, the C-H bond in methane was calculated to use sp2.21 (sp3.35 using overlap densities) orbitals. Several conclusions resulted: i) hybridization in lower-symmetry molecules is frequently negligible; ii) hybridization might be most in evidence in first-row hydrides; iii) sp hybridization is much more in evidence in bonds formed by elements to the left of the periodic table. “The utilization of s and p orbitals in bonding, as estimated from gross atomic population data and overlap density data, varies so far from the familiar spn stereotypes that there is no justification for retaining the sp-ratio/bond-angle rule in its usual form” [110]. Furthermore, “The Walsh-Bent hypothesis, that the attachment of electronegative groups favors the use of p rather than s orbitals in bonding by a central atom, is not supported” [111].
In 1984, further doubt was expressed about the quantum-chemical calculation of hybridization. “Modern quantum chemistry, however, has no real place for [hybridization], and many theoretical chemists regard [hybridization] as problematic or as outdated…In the molecular-orbital (MO) approximation, there is no need for ad hoc assumptions about hybridization. In fact, it is possible to manage without this concept at all.…This applies particularly to the concept of hybridization [in higher main-group elements], which should be viewed with considerable caution” [112].
During the next decade, quantum-chemical calculations were directed to determine the ‘best’ hybrids for unsaturated carbon compounds – the s-p model or the tau model. There were previous studies on this topic, but period 1970-1990 was particularly critical as software and computational power increased. Making the first self-consistent, correlated SOPP-GVB calculations for ethene and ethyne in 1972, Goddard et al. concluded that the s-p model was a superior description [113].
In 1986 Palke restated that in Hartree-Fock [HF] methods both models are equivalent. “Because all three are merely unitary transformations of one another, they have the same energy and the same total charge distribution; The HF method itself provides no criterion for preferring banana bonds to s plus p bonds.” Using a valence-bond wave function fully optimized as an LCAO function using a non-orthogonal orbital method for ethene, however, supported the lower energy of bent bonds but with an unexpected mix of orbitals. “Each of the orbitals in the wavefunction is sp1.37d0.07 and has 24% p character.” Furthermore, “Our preliminary results using the method for the acetylene molecule indicate that a wave function consisting of equivalent hybrid bonds (three pairs in that case) is lower in energy than a s plus two p bond wave function” [114].
Messmer and Schultz also supported the tau model for CO2 [52], difluoroacetylene [115] and benzene [116]. “Results for the carbon-carbon triple bond suggest that such bonds may be better described in terms of ‘bent bonds’ than by the traditional combination of σ and π bonds” [115]. In subsequent, more comprehensive papers, the same authors used self-consistent full GVB wave functions on small molecules, including ethene and ethyne; they concluded, “Our results yield bent bonds as the favoured bonding description, showing that the σ, π bond descriptions of multiple bonds are artifacts of approximations to the full independent-particle equations” [117]. The hybrids calculated using full-GVB functions for ethene (sp1.84, 85° angle between orbitals) and ethyne (sp1.64, 115° angle) show conflicting results. In a second paper, the authors showed that the perfect-pairing spin-coupling restriction in GVB functions is appropriate for multiple bonds [118]; a third paper concluded that this calculation extends to conjugated systems, such as benzene. “Based on energetic considerations, the bent-bond model serves as a better framework with which to describe the electronic structure in systems exhibiting resonance than the σ, π bond model” [119].
Based on the spin-coupled (SC, also known as full-GVB) point of view, as opposed to the one-configuration wave-function Hartree-Fock (HF) and valence-bond (VB) methods, ethene and ethyne were calculated with a high-level triple-zeta valence basis set with polarization (TZVP); Karadakov et al. determined that the energy difference between the models was negligible. “Thus, from an energetic point of view, both constructions provide an equally good starting point for the treatment of correlation effects beyond the one-configuration approximation” [120]. In a subsequent paper by the same authors, using a standard active space, which included all valence electrons, instead of a minimal active space, they observed an even further decrease in the energy separation between the two bonding models [121].
All these calculations to determine the best hybrid-orbital model are perhaps moot. “No experiment can possibly distinguish between a s, p double bond and double bent bonds in any system, and therefore neither can be proven to be ‘right’ in an absolute sense; both are approximate descriptions” [117].
In the calculation of several simple molecules using level HF-SCF/6-31G*, no support of any core assumption of hybridization was observed [122]. A model with non-bonded interactions was preferred to explain trends in molecular geometry.
In another attempt to determine whether hybridization exists in methane using quantum-chemical calculations at a high level, both RHF/TZVP and B3LYP/TZVP models failed to show any quantitative evidence of sp3 HAO in either coordinate space or momentum space [123].
Using a natural-bond-orbital (NBO) analysis, Alabugin et al. provided several examples of hybridization effects pretending to control the structure and reactivity [124]. In reality, the calculations showed simple correlations between the NBO hybridization values and selected properties, but correlation is not causation.
In a valence-bond approach to hybridization, Shaik et al. [125] adopted a critical view of the initial step in the hybridization process, the promotion of electrons in the carbon atom; state 5S lies well above the ground state of the carbon atom by 402 kJ/mol. Second, they questioned the orthogonality on proposing partial or overlapping hybrids: “Indeed, why should the HAO be constrained to be orthogonal to each other? Modern ab initio valence-bond (VB) methods neither apply this constraint, nor do they require orthogonality” [125]. Third, calculations on methane and ethyne produced surprising results. For methane with a full 81-structure valence-bond self-consistent-field [VBSCF] wave-function calculation in a 6-31G(d) basis set, a sp1.76 hybrid was considered optimal. A 27-structure VBSCF calculation for ethyne provided a sp0.41 hybrid [125].
In a series of recent papers [126–128] Dunning and co-workers provided the most nearly complete and advanced computational analysis of hybridization of carbon. The first paper provided spin-coupled generalized-valence-bond [SCGVB] calculations on methane, ethane, ethene and ethyne using basis set aug-cc-pVQZ, but did not focus on hybrids. Note that GVB theory is referred to as SCGVB theory as a full GVB wave function is equivalent to the spin-coupled valence-bond (SCVB) wave function. This method was suggested to provide future computational evidence of hybridization: “GVB theory is also able to provide invaluable insights into one of the most compelling questions in chemistry: How are atoms changed by molecular formation?” [126]. In a second paper, these authors investigated SCGVB calculations in the series in Group 14 elements: CH4, SiH4 and GeH4 [127]. For methane using aug-cc-pVQZ basis sets for C and H, hybridization sp0.6 was calculated. The authors concluded, “Each of the X atom bond orbitals in XH4 is pointed toward the hydrogen atom to which it is bonded.…This is so in spite of the fact that the X atom bond orbitals are not sp3 hybrids. Thus, the tetrahedral structure of the XH4 molecules is not a result of the sp3 hybridization of the X atom orbitals” [127]. In their third paper, Dunning et al. used SCGVB equations with basis sets aug-cc-pVQZ and aug-cc-pV5Z for methane and aug-cc-pVQZ for calculations on ethene and ethyne for the carbon and hydrogen atoms, testing the s-p description of the bonding in C2H4 and C2H2 favored by organic chemists [128]. The abstract in this paper states: “It is now clear that the orbitals in modern valence-bond wave functions do not follow the hybridization rules of traditional valence-bond theory. These findings imply that, in modern valence-bond theories, other factors are responsible for the structures and properties of molecules that are traditionally attributed to orbital hybridization.” The calculated hybridization at the highest level (quintuple-zeta quality) indicated sp0.56 hybridization for methane. Ethene had calculated C-H bond hybrids sp0.57 and C-C hybrids sp0.39; ethyne presented C-H bond hybrids sp0.76 and C-C hybrids sp0.37. No such ‘hybrid’ corresponds to the classical viewpoint (i.e. sp3 for methane, sp2 for ethene, sp for ethyne). For all organic molecules, “a strong bond can be formed with the electrons in the (2s–, 2s+) lobe orbitals as well as the 2px and 2py orbitals. This accounts for the tetravalence of the carbon atom—there is no need in SCGVB theory to invoke the high-energy C 5S state or a predetermined hybridization ratio” [128].
In 2021, Bickelhaupt et al. claimed that the tendency of bond length to increase from alkynes to alkenes to alkanes might be explained by steric strain (Pauli repulsion) instead of hybridization. Their analysis of ethane, ethene, ethyne, propane, propene and propyne based on Kohn–Sham molecular-orbital theory (DFT), and energy-decomposition analysis (EDA) shows minimal traditional hybridization effects on bonding. “Our energy decomposition analysis as a function of the C-X (X = H, CH3) distance shows that, in contrast to present-day textbook knowledge, the orbital interactions DEoi are not responsible for the stronger and shorter sp-hybridized C-H and C-C bonds” [129].
We doubt that any future calculational studies can achieve correspondence between the model of traditional hybridization used in organic chemistry and the quantum-chemical world. We predict that future results will move away from HAO and hybridization and will use other data to predict chemical properties and reactivity.
7) Evolving Models: 1982-1999
During this period, a flood of support of the hybridization model arrived in textbooks, research articles and computer analysis. We regard that ‘tribute’ articles during this period are historically necessary to include but that detailed description is unnecessary.
The ‘special hybrid orbitals’ sp, sp2 and sp3 were reviewed and extended to ‘general hybrid orbitals’ by Bingel and Lüttke. With this extension, the special assignments could be applied to most symmetry groups without experimental support. “To clarify the concepts, it may be mentioned at this point that, for example, the designation sp2 does not indicate, as is often incorrectly done, only the three special trigonal hybrid orbitals, but also those three general hybrid orbitals, for the construction of which in total one s and two p AO’s are used. The distribution of, for example, the s AO over the three hybrid orbitals is uniform in the special case (with 33 % each), but it is non-uniform in the general case” [130]. This trigonometric system was extended and programmed into software for calculation [131].
Hermann wrote a review of hybridization as a fifty-year tribute to the original paper by Pauling [132]. Another paper dedicated to Pauling on his 85th birthday thoroughly discussed orbital hybridization and symmetry [133]. This review was followed in August 1988 by volume 169 of Journal of Molecular Structure: Theochem dedicated to Linus Pauling and to the latest developments in hybridization, approximately sixty years after its development. Although a full account of all the papers in this issue is impractical, we provide a table (Table 2) with the titles and authors of the articles about hybridization presented in this volume.
Table 2. Index of selected articles about hybridization in Journal of Molecular Structure, volume 169, 1988


8) A New Century: 2000-2021
The twenty-first century brought a new generation of chemists to review the contribution of hybridization to chemistry. As in most sciences, the pendulum of truth swings from complete acceptance to critical rejection; this change should be documented as a success of the maxim that scientific truth is never settled; skepticism and dissent, not consensus and authority, define chemistry.
Gil published a book Orbitals in Chemistry that included a section on the Use and Misuse of the Hybrid Orbital Concept. In that section, he warned against using HAO as an explanation – there is no cause-effect relation between HAO and geometry; the two manifestations – HAO = mathematical, and geometry = physical – should not be conflated. HAO cannot be considered a physical phenomenon. “No geometric parameter or any other molecular property can be explained by invoking hybrid orbitals.” For example, the interpretation of bond energies, bond lengths, force constants, bond polarity, acidic character, molecular energy etc., should not be attributed to hybridization [134].
In 2000, Barbier and Berthier recounted a half century of hybridization in a review dedicated to Del Re. Interestingly, they indicated that for the original concept of hybridization, “If we pass over the questions connected to its theoretical origin, hybridization can be considered as a simple way for describing, both in speaking and in writing, the so-called molecular observables in terms of atomic components… i) it is a way for constructing ‘chemical orbitals’, that is to say, wave functions which preserve the concept of bond properties and in the same time gives us good starting points for the development of various semi-empirical or ab initio treatments. ii) it is not an experimentally observable phenomenon, but according to Coulson merely ‘a feature of a theoretical description’.” [135]. The review then focused on a procedure of overlap-matrix localization ab initio pioneered by Del Re.
Boeyens in a series of published works after the turn of the century, presented his strong arguments against hybridization. In New Theories for Chemistry (2005), he presented an anomaly in orbital momentum, a lack of prediction or explanatory power of HAO and a problematic rotational barrier in ethene as limitations of hybridization. “The functions px, py, pz do hence not represent states that can co-exist on an atom and linear combinations of real functions, such as sp2 and sp3 have no quantum-mechanical meaning.” [136].
In his next book, Models, Mysteries and Magic of Molecules (2008), he tried to provide a critical perspective about hybridization and provided three logical arguments against their use. The first attack claims that px, py and pz orbitals can never occur together and can never be combined in linear combinations because at least two form an orthogonal complex pair. Another problem is that these orbitals must share the same quantum numbers. “a more serious objection is that each of them has the same magnetic quantum number m = 0. In addition, they also have n = 2 and l = 1. The assumption therefore violates the Pauli exclusion principle. However, the sp3 carbon has three electrons with n = 2, l = 1, and m = 0, and there are only two possible spin values, ms = ±½. The exclusion principle is widely recognized to be as ruthless as the second law of thermodynamics. The idea of sp3 hybridization is therefore as ludicrous as perpetual motion.” His third critique is that orbital momentum is not conserved. “In hybridization theory, this conservation of angular momentum is ignored. Despite the large difference in energy of the s and p-states, linear combinations of the four eigenfunctions are nevertheless assumed to produce alternative solutions to the wave equation, which define the allowed geometries of carbon compounds. This act of faith is supported neither by the laws of physics nor by the mathematical model.” [137].
In 2008, Boeyens continued his attack in another book in which he criticized the entire hybridization process. “A further new theory that developed to overcome this problem is known as the theory of orbital hybridization. In order to simulate the carbon atom’s basicity of four, an additional orbital is clearly required. The only possible candidate is the 2s orbital, but, because it lies at a much lower energy and has no angular momentum to match, it cannot possibly mix with the p-eigenfunctions on an equal footing.” Continuing, he reiterated his previous arguments: i) orbitals px and py are not equivalent to pz – they are identical to it, in a rotated state; ii) the disregard of the Pauli exclusion principle (same set of quantum numbers); iii) the disregard of the conservation of orbital momentum as s is promoted to p. “Not only is hybridization an artificial simulation without scientific foundation, but even the assumed ‘orbital shapes’ that it relies upon are gross distortions of actual electron-density distributions” [138].
In a subsequent book chapter about quantum mechanics, he eliminated two more assumptions about hybridization. As HAO are wave functions, their squares should provide information about the electron density – they do not! “However, if we take the square of any tetrahedral orbital, constructed according to the principles of Pauling, we find that (sp3)2 = (s+px+py+pz)2 represents a spherical distribution because all cross products disappear due to the mutual orthonormality of the atomic orbitals. The inevitable conclusion is, therefore, that tetrahedral orbitals do not represent preferred directions of bonding in space as stated by Pauling, but merely restate the sphericity of atoms in another way. The same holds for the squares of all hybridization types, thus merely reaffirming the statement that free atoms are spherical objects” [86]. Another critique presented against orthogonality removed any justification for the continued use of HAO: “this visualization of chemical interaction is based on a false picture. It is a mathematical property of spherical harmonics that they constitute an orthogonal set. This requires that the 2s and 2pz functions be orthogonal and hence have zero overlap. The seductive picture of an sp hybrid is therefore ruled out by the orthogonality condition” [86]. In 2013, he concluded his objections to hybridization on exposing the cyclic-argument nature of the process; “No amount of hand-waving can circumvent this conclusion. The elaborate procedure whereby these orbitals are incorporated in further ‘hybridization’ to define the combinations sp3, sp2 and sp to simulate tetrahedral, trigonal and linear sets of orbitals is likewise without quantum-mechanical meaning. At best, it amounts to a classical reconstruction of these geometries” [139].
Finally, in The Quantum Gamble (2016), published just before the death of the author, he questioned the continued support of an obsolete model. “At the present time one marvels at the pathological reluctance to abandon the discredited model of orbital hybridization, and linear combination of atomic orbitals as an explanation of chemical interactions.…A bewildering aspect of this situation is to find well informed people refusing to take serious note of this argument against hybridization. They respond with rebuttals such as ‘quantum numbers are not required in this instance’, or ‘the exclusion principle should not be taken too literally’, even ‘with a proper understanding of quantum theory there is no problem’ ” [140].
The authors of this review have provided thirteen reasons, both logical and practical, for the removal of HAO in organic chemistry [1, 2]. We provided also a plan to remove HAO completely from introductory chemistry. The list of items indicated in these two papers is summarized below.
- The formation of four real tetrahedral HAO using linear combinations of real and imaginary parts in spherical polar coordinates is mathematically impossible and logically unsound.
- The combinations of HAO in various sets, such as the set described as sp3 were devised to generate tetrahedrally oriented hybrid functions; their subsequent use to explain a tetrahedral structure of methane is manifestly a circular argument.
- To attribute the structure of methane to sp3 tetrahedral hybrid atomic orbitals (neglecting an alternative description, above, as sp2 tetrahedral hybridization) is at least an exaggeration and a grossly misleading simplification.
- Trigonal hybrid functions (to which reference is sometimes made as sp2 but which are distinct from the tetrahedrally oriented sp2 hybrid functions specified above) suffer from the same unjustified discard of √−1 as a coefficient of py.
- If one undertakes a molecular-orbital calculation for CH4 according to a standard quantum-chemical procedure with a basis set comprising only four 1s functions on H, and on C (implicitly involving only 2s and 2p functions), one obtains exactly the same structure of CH4 and the same energy as modern valence-bond calculations.
- Those solutions of Schroedinger’s equation in spherical polar coordinates as presented above are applicable to only an atomic system with rigorously spherical
- There is neither necessity nor justification for hybridization as a consequence of quantum mechanics.
- Describing a) an atomic center in a molecule or b) a lone pair or c) a bond as a hybrid is unacceptable.
- One cannot claim hybridization as the cause of an observation or physical property.
- No direct experimental evidence for HAO exists, and can never be found.
- HAO constitute a flawed model − flawed mathematically, flawed practically and flawed pedagogically.
- Applying sp3, sp2 and sp HAO is not generally applicable to all organic molecules.
- How can one easily understand systems in which where the bond angle is less than the minimum hybrid bond angle, 109.5°?
These thirteen reasons, the recent quantum-chemical results of Dunning, the critiques of Gil and of Boeyens, the totality of evidence from the twentieth century, signal the end and defeat of a discredited model. The cumulative evidence in the quest for truth has been confirmed in several scientific fields – chemistry, physics and mathematics – and several approaches to knowledge – philosophy, empiricism, logic and pedagogy. There is no need to continue to use the hybridization model, no need to teach HAO in introductory chemistry classes and no need to propagate the language of hybridization as dogma in universities.
9) Conclusion
One must occasionally peer into the past to see into the future. This review provides a snapshot over almost a century of the discovery, growth, evolution and denunciation of hybrid atomic orbitals. At every stage, progress has been made, and will continue to be made, to understand chemistry. Perhaps after 100 years, we can accept that the time has come for hybridization to be discarded. The hybrid atomic-orbital model (sp3 & sp2 or tau or non-integral hybrids) can be summarized in the following indisputable statements.
- There is no mathematical basis for hybridization.
- There is no quantum-mechanical basis for hybridization.
- There is no modern quantum-chemical (computational) evidence for hybridization.
- There is neither physical nor experimental evidence for hybridization (nor can there ever be).
- There is neither linguistic nor mnemonic nor practical reason for the use of hybridization.
- There is valid explanation of neither molecular structure nor property nor reactivity nor spectra using hybridization
- There is no pedagogical reason for the discussion of hybridization (except in an historical or mathematical context).
HAO cannot be considered today a tenable scientific idea, reality, fact, model or theory; they do not exist.
Will chemistry collapse without the hybridization model? Obsolete models have been previously eliminated for the advance of science. This model might continue to be used in advanced research papers, even though it is irrelevant for chemistry, physics and material science. It might continue to serve as a model for basis sets in quantum chemistry, perhaps under another name.
)