Document Type : Review Article
Authors
1 Department of Chemistry, MMEC, Maharishi Markandeshwar (Deemed to be University), Mullana-133207, Ambala, India
2 Department of Applied Science, UIET, Kurukshetra University, Kurukshetra – 136119, India
3 Department of Chemistry, Kurukshetra University, Kurukshetra – 136119, India
Abstract
Determination of palladium (II) in traces is significant due to its widespread applications in various fields, especially as alloys in ornaments, electrical appliances and significantly as a catalyst for various synthetic reactions in chemistry. Determination is additionally vital due to the hazardous ecological impacts of the element on biological networks. Thus, attributable to the cordial and poisonous nature of the element, there has been an extensive interest in the detection and determination of its content in various natural and industrial samples. Numerous analytical techniques are accounted for analysis of the metal ion. However, UV-Visible spectrophotometry has been much favorable because of its simplicity, rapidity, inexpensiveness, sensitivity, selectivity, and precision. The present article refreshes ongoing advancements for analysis of Pd (II) spectrophotometrically and fundamental states of the technique required for micro determination of elements in the samples of analytical interest.
Graphical Abstract
)
Keywords
Main Subjects
- Introduction
palladium (Pd), a chemical element having atomic number 46 together with platinum (Pt), rhodium (Rh), ruthenium (Ru), iridium (Ir) and osmium (Os) creates a group called PGMs. The metal is soft silvery-white having the least density and melting point out of other PGMs. It is ductile in nature and stable towards oxygen at standard temperature. It exists in seven isotopic forms, with 107 Pd being the most stable radioisotope having a half-life of 6.45 years.
Together with other platinum group metals, palladium is found both in primary and secondary deposits which contain the world's largest known reserves of platinum metals. Secondary or placer deposits result from erosion or weathering in the Norine belt of the Bushveld igneous complex in the Transvaal and the Sudbury district of Ontario and Canada. Some essential minerals of palladium are also palladium, eugenesite, braggite, potarite, and clausthalite. The estimated proportion of palladium in the earth's crust is 8.5 × 10-13, 1.9 × 10-5 in meteoric iron and 4.5 × 10-6 in troilite. The presence of palladium in the sun's photosphere was reported by J.N. Lockyer [1].
1.1 Palladium usages
The most extensive use of palladium is as an alloying element. Its alloys with copper and silver are used in electrical contacts because of low electrical conductance. In small amounts the metal also finds utility in cast dental alloys of dental amalgam to decrease corrosion and increase the metallic luster of the final reinstatement [2]. Addition of palladium increases the passivity of stainless steel. A small addition of palladium, about 0.1 % to titanium provides resistance towards boiling acid solutions. Palladium on alloying with gold protects electrically heated furnaces from overheating and finds application in astronomical instruments. A moderate amount of palladium is used in making jewellery called white gold a gold alloy decolorized palladium addition.
Another essential use of palladium is to behave as a catalyst in hydrogenation and dehydrogenation reactions. Palladium is essential for Lindlar catalyst, also known as Lindlar's Palladium [3] and also an effective carbon-fluorine bonds catalyst [4]. The auto-oxidation of 2-acetylanthraquinol using a palladium catalyst is used in the industrial synthesis of H2O2. Palladium is also used in hydrogen storage as it readily absorbs hydrogen at room temperature and forms palladium hydride having formula PdHx with x < 1 [5]. Palladium is also used as a coating for mirrors in searchlights.
An idea of some elements, which are generally associated with palladium in natural and industrial products, is presented in Table 1.
Table 1: Some natural and industrial palladium products

1.2 Health and environmental effects of palladium
Palladium may cause skin, eye, or respiratory tract disturbance. The fluid may cause burns to the skin and eyes. Palladium is viewed as of low poisonous quality inadequately consumed by the body. When ingested, inhaled, or absorbed via the skin, all palladium compounds are extraordinarily hazardous and cancer-causing. It harms the liver, bone marrow, and kidneys [6]. In fragile people, small quantities are enough to trigger hypersensitive reactions. Miners, dentists, and chemical workers are among the workers who have been exposed to palladium. The latter group is mainly exposed to palladium salts, some of which can cause severe skin and eye problems. Palladium can be ingested by the general public, mainly through mucosal contact with dental reconstructing attempts and palladium-containing stones, conceivably by means of emanations from Pd catalysts.
- Determination of palladium
Due to the occurrence in low concentrations of palladium in many natural samples, the enrichment in the by-products of many industrial processes, use in many palladium alloys in combination with other transition elements, as catalysts. Many health and environmental effects put a considerable interest in the methods of detection and determination based on sensitive and specific reactions of the metal.
A number of methods of determination of palladium have been published in the past including gravimetric [7, 8], titrimetric [9], AAS [10], X- ray fluorescence [11, 12], NAA [13, 14], ICP-ES [15], ICP-MS [16], solvent injection method [17, 18] and UV-VIS spectrophotometry.
It is clear that the interference of a high number of metal ions substantially affects the gravimetric and volumetric procedures used to determine palladium. NAA, AAS, ICP-ES, ICP-MS, and X-ray fluorescence procedures are highly sensitive for palladium trace measurement, but require more expensive equipment and initial concentration and separation of the element. Palladium's rising use as an alloying element, a catalyst and its environmental consequences highlight the urgent need for research into its determination methodologies.
Hence, of the several approaches that may be used to determine the metal in traces, UV-VIS spectrophotometric methods are the Palladium (II), most widely utilized because they need less instrumentation and methodology. For micro-level determination of Palladium (II), spectrophotometric methods are considered quite convenient and frequently used for the same reason. In the presented review article an update on the spectrophotometric determination methods of palladium using different complex-forming or chelating agents have been searched. The methods are quite advantageous from an analytical point of view of the metal ion. Depending upon the nature of complexing conditions, the methods have been discussed under three headings.
2.1 Liquid-liquid extraction technique
Liquid-liquid extraction (Figure 1) or solvent extraction or partitioning is a technique for separating metal complexes based on their relative solubility in two immiscible liquids, typically the polar aqueous or non-polar, non-aqueous organic solvents resulting in a net transfer of the metal complex from one liquid phase to another. The technique is highly fruitful in developing the extremely sensitivsusceptible and selective methods of determining the transition elements, including palladium (II), as mentioned in the subsequent-subsequent discussion.
In modified cold-induced aggregation micro extraction, 1-(2-pyridylazo)-2-naphthol [19] was utilized as a complex-forming agent in a micro extraction approach based on ionic liquids to detect palladium in saline solution. The extraction of metal ions from water samples was discovered to be rapid and straight forward. The detection limit was 0.0004 µg mL-1 under ideal conditions and the relative standard deviation calculated for five replicates was 2.23 %. The compound obeyed linearity between 0.005-0.1 µg Pd (II) mL-1.

Figure 1. Liquid-liquid extraction technique
Pd (II) reacted with 4-(2-pyridylazo)-resorcinol (PAR) [20] to form a red complex at 90°C in the pH range 9.0-11.0. The complex was quantitatively extracted into molten naphthalene and exhibited wavelength maximum at 520 nm in naphthalene-chloroform where the reagent blank absorbed negligibly, Beer's law range, molar absorptivity and sensitivity of the formed complex were 0.1-2 µg Pd (II) mL-1, 8.0 × 105 L mol-1cm-1 and 0.00049 µg cm-2, respectively. The inclusion of EDTA effectively concealed the significant influence of Co (II), Fe (II), and Bi (III).
4-(5-Chloro-2-pyridine)-azo-1,3-diaminobenzene (5-Cl-PADAB) [21] Pd (II) reacted to form a colored compound in 1.8 M H2SO4 medium giving maximum color intensity at 570 nm. The complex showed linearity in the range of 0-1.2 mg Pd (II) L-1 with a molar extinction coefficient of 6.38 × 104 L mol-1 cm-1. Out of 30 ions studied for interference, Ag (I), Pb (II), and Cr (VI) interfered seriously with the determination. However, the interference was masked either by the precipitation or reduction method. In a 3.0 M H3PO4 medium over a water bath at 100° C, Pd (II) reacted with 4-(5-chloro-2-pyridyl) azo-1,3-diaminobenzene [22] to form stable 1:2 complex obeying Beer's law range up to 1.2 mg Pd (II) L-1. RSD of the method was < 4%.
At pH 9, 2-[(E)-N-(2-{[2-[(E)-[(2-hydroxyphenyl) methylildene] amino] phenyl} (methyl) amino} phenyl) carboximidoyl] phenol [23] formed a 1:1 complex of Pd (II), quantitatively extracting in chloroform and exhibiting maximum absorption at 560 nm. Molar absorptivity as evaluated was 0.47 × 102 L mol-1 cm-1. The extractive spectrophotometric method was satisfactorily applied to Pd (II) detection from several mixtures.
2-(5-Carboxy-1,3,4-triazolyl-azo)-5-diethylaminobenzoic acid [24] formed a stable purple coloured complex of Pd (II) in the ratio one to two (M to L) which exhibited absorption maximum at 548 nm; apparent molar annihilation coefficient and Beer's law range as noted being, respectively 9.32 × 104 L mol-1 cm-1 and 0.08-0.8 mg Pd (II) L-1.
Pd (II) was spectrophotometrically determined after its complex formation with 3-hydroxy-3-propyl-1-(4-carbamimidoylsulphamoyl) phenyltriazine [25] (Scheme 1) in an alcoholic medium at a pH 1.8-2.2. The 1:2 violet coloured complex absorbed maximally at the wavelength 367 nm possessing molar absorptivity of 8.372 × 103 L mol-1 cm-1.

Scheme 1. 3-Hydroxy-3-propyl-1-(4-carbamimidoylsulphamoyl) phenyltriazine
The complex formation reaction between Pd (II) and 4-amino-3-mercapto-6-methyl-1,2,4-triazine (4H)-5-one (AMMT) [26] had been studied where a light yellow stable (at least 24 h) complex was formed between the two at pH 11. The 1:2 complex exhibited maximum colour intensity at 384 nm with the linearity exhibited up to 10.4 mg Pd (II) L-1 with molar absorptivity 2.1121 × 104 L mol-1cm-1. Mg (II) and Zr (IV) interfered seriously with the determination.
For the spectrophotometric detection of Pd, another triazine derivative, 1-(2-naphthalene)-3-(2-thiazole)-triazine (NTT) [27], was used as a new chromogenic reagent (II). The procedure was straightforward, selective, sensitive (ε = 4.07 × 104 L mol-1cm-1) and linear up to 1.75 µg mL-1 Pd (II).
A brown complex of Pd (II) was formed with a new triazine derivative, 1-(4-antipyrine)-3-(2-thiazolyl) triazine (ATTA) [28] in the presence of organophosphate (OP) and Na2B4O7-NaOH buffer solution of pH 9.5. Linearity was up to 0.01-1.60 µg mL-1 Pd. The method was highly sensitive, having a molar extinction coefficient of 5.33 × 104 L mol-1cm-1.
From a 0.0-5.0 M sodium malonate medium in 25 ml aqueous volume with 1:1 ammonia, Pd (II) was quantitatively extracted into xylene after reacting with N-decylpyridine-4-amine [29] (Scheme 2). The developed method was sensitive with a molar absorptivity and sensitivity of 1.9 × 105 L mol-1 cm-1 and 0.065 µg Pd(II) cm-2, respectively at 410 nm.

Scheme 2. N-Decylpyridine-4-amine
A simple, fast and sensitive (ε = 5.320 × 103 L mol-1 cm-1) procedure had been developed for spectrophotometric investigation of Pd (II) using 2-hydroxy-5-methylacetophenoneisonicotinoylhydrazone [30]. The 1:1 yellow complex was extracted into chloroform from 0.010 - 0.015 M H2SO4 medium, showing λmax of 385 nm and linear range 2.0-9.0 mg Pd L-1.
Pd (II) was extracted quantitatively into toluene following treatment with isonitroso-p-methylacetophenonephenylhydrazone in an aqueous solution of pH 0.0-5.0 and 0.1-1 M solution of acetic acid and mineral acids in the presence of 1 mL of 2 M sodium acetate [31]. At 470 nm, the extracted product had a molar absorption value of 1.3305 × 104 L mol-1 cm-1. The 1:2 complex was shown to be stable for at least 48 h at RT and followed Beer's law over the Pd (II) concentration range 0.1-10 µg mL-1.
2-Aminoacetophenone isonicotinoyl hydrazone [32] was used as a sensitive (ε = 3.00 × 104 L mol-1 cm-1) and selective reagent for spectrophotometric detection of Pd (II) where it formed a 2:1 (L:M) intense orange-red complex in the acidic medium of pH 3-5. The complex obeyed linearity up to 3.00 µg Pd (II) mL-1 at pH 4.
p-[N, N-bis (2-chloroethyl) amino] benzaldehyde thiosemicarbazone [33] (Scheme 3) was used as a new analytical reagent for the spectrophotometric detection of Pd (II). At a pH of 1.0-2.0, a 1:2 yellow complex was formed between the two (with HCl). After combining the reagents, the formation took 5 min to reach maximum color development and remained stable for at least 2 h at 60 ℃. The linear calibration graph was followed up to 2.64 µg Pd (II) ml-1 with a molar absorptivity 4.05 × 104 L mol-1 cm-1. Cu (II) with Pt (IV) interfered seriously.

Scheme 3. p-[N,N-bis(2-chloroethyl)amino]benzaldehyde thiosemicarbazone
From an aqueous condition of pH 0.0-4.0, Pd (II) was quantitatively extracted into chloroform after its complex formation with isonitroso p-nitroacetophenone thiosemicarbazone (HINATS) [34]. The 1:2 chloroform extract of M:L complex absorbed maximally at 410 nm and obeyed linearity at 5.0-80 µg Pd (II) mL-1 with stability for 2 days at ambient temperature and the extinction coefficient of 9.10 × 102 L mol-1 cm-1. Procedure found satisfactory application to Pd (II) determination in catalysts and alloys.
Pd (II) was determined spectrophotometrically using 4-(N, N-diethylamino) benzaldehyde thiosemicarbazone (DEABT) [35]. In an aqueous solution, the 1:2 weakly soluble yellow complex was produced, which dissolved fully in DMF-Ethanol. However, in a potassium hydrogen phthalate-HCl buffer media, a maximal and stable value of absorbance was reached in the pH range 2.6-3.4. After being kept for 5 minutes to enable maximal colour development, the complex absorbed the most at 408 nm and stayed steady for around 2 hours. Lnearity was valid up to 3.60 µg Pd (II) mL-1 with the molar absorptivity of 3.33 × 104 L mol-1 cm-1; Cu (II) and Pt (IV) causing significant interference. Palladium was successfully determined in Pd-Rh and Pd-Ni alloys, catalysts and variable synthetic mixtures.
At pH 4.0, Pd (II) and N-Ethyl-3-cabazolecarboxaldehyde thiosemicarbazone [36] reacted to generate a yellowish-orange 1:1 chelating complex. The compound could be extracted with n-butanol and at 410 nm had the highest color intensity. The compound had a Beer's law range 0.0-6.6 µg Pd (II) mL-1 and molar absorptivity of 1.647× 104 L mol-1 cm-1. The approach successfully determined Pd (II) levels in water samples, synthetic mixes, and hydrogenation catalysts.
Pd (II) was generated in a 1:1 yellowish-orange complex with 2,6-diacetylpyridine-bis-4-phenyl-3-thiosemicarbazone [37] (Scheme 4) in an acetic acid - sodium acetate buffer solution at pH 3.5 - 4.5 that was extractable into isoamyl alcohol, had λmax at 410 nm and was stable for 46 h. With the molar absorptivity of 1.156 × 104 L mol-1 cm-1, Beer's law was applicable in the Pd (II) concentration range of 0.0-12.6 µg mL-1 .

Scheme 4. 2,6-Diacetylpyridine-bis-4-phenyl-3-thiosemicarbazone
In an acetate buffer solution of pH 4.0-6.0, benzyloxybenzaldehyde thiosemicarbazone [38] (Scheme 5) was generated and employed for the extractive spectrophotometric measurement of Pd (II), producing a highly selective and stable (stability > 48 h) 1:1 yellow complex. The compound was extracted into cyclohexanol and measured at 365 nm for maximum colour intensity. The complex's Beer's law range, molar absorptivity, and relative standard deviation were 5 - 60 mg Pd (II) L-1, 0.4 × 104 L mol-1 cm-1 and 0.321%. The determination was substantially hampered by Cu (II). On the other hand, the approach was used to determine palladium in water samples and synthetic mixtures.
In methanol, 5,6-diphenyl-2,3-dihydro-1,2,4-triazine-3-thione [39] formed 1:1 yellowish-orange complex of Pd (II). As observed, optical and statistical parameters were: λmax, linearity range, molar absorption coefficient and RSD as 385 nm, 10-50 µg Pd (II) mL-1, 6.67 × 103 L mol-1 cm-1 and ≤ 3.96%, respectively.

Scheme 5. Benzyloxybenzaldehyde thiosemicarbazone
In a 0.5-3.0 M HNO3 medium N,N,N',N'-tetra(2-ethylhexyl)thiodiglycolamide [40] (Scheme 6) interacted with Pd (II) to form a 2:1 yellow complex, extractable into the neutral solvent n-dodecane having a maximum colour intensity of 300 nm. The colored complex obeyed linearity in the range 1.0-15.0 µg Pd (II) mL-1 with the evaluated molar absorptivity of 1.29 × 105 L mol-1 cm-1 and relative standard deviation as < 0.5%.
Pd (II) produced a 1:1 complex with 4-(4-methoxybenzylideneimino)-5-methyl-4H-1,2,4-triazole-3-thiol [41] in HCl medium, which was extractable into chloroform and had a reproducibility of 99.00 ± 0.95 %. Similarly, 4-(4-ethoxybenzylideneimino)-5-methyl-4H-1,2,4- triazole-3-thiol [42] reacted with Pd (II) to give a simple and rapid liquid-liquid extraction method with RSD=0.95%.
 Scheme 6. N,N,N',N'-tetra-(2-ethylhexyl)thiodiglycolamide
Scheme 6. N,N,N',N'-tetra-(2-ethylhexyl)thiodiglycolamide
Based on the interaction of metal ions with 5-hydroxyimino-4-imino-1,3-thiazolidin-2-one [43] at pH 5.0, a new, simple, and rapid spectrophotometric approach for determining palladium was used in the investigation of intermetallics Yb40Pd38Sn22 and Yb40Pd40Ga20. The 1:1 complex absorbed light at 350 nm and had the ε value 5.9 × 103 L mol-1 cm-1. The compound was linear in the palladium concentration range 6.0 × 10-6 – 6.0 × 105 molar.
Pd (II) formed a stable (stability > 70 h) 1:1 yellow coloured complex with o-methylphenylthiourea [44] in 0.8 M HCl which was extracted 100% in chloroform and had λmax at 340 nm. Beer's law range and molar extinction coefficient evaluated were 0.01-15.0 µg mL-1 of Pd (II) and 2.85 × 103 L mol-1 cm-1. Palladium from multi-component mixtures and hydrogenation catalysts was analyzed using this method.
Quantitative extraction of a 1:2 complex of Pd (II) with 2-(5-bromo-2-oxoindolin-3-ylidene) hydrazine carbothioamide [45] was done into n-amyl alcohol from 0.1-3.0 M HCl medium of pH 0.0 - 4.0. An intense peak of the extracted complex was noticed at 520 nm. 1.0 – 30.0 µg Pd (II) mL-1 and 7.450 × 103 L mol-1 cm-1 were respectively the linearity range and molar absorption coefficient of the formed complex at 520 nm. Pd (II) in Pd-Charcoal and Pd-BaSO4 catalysts were successfully analyzed using the proposed approach.
The liquid-liquid extraction behavior of Pd (II) was studied as its 2-hydroxy-1-naphthalene carboxaldehyde hydrazine carboxamide complex [46] (Scheme 7). The 1:2 orange complex was quantitatively extracted into ethylacetate at pH 4.2 with an absorption maximum at 410 nm. In ethyl acetate, the extracted complex was stable for 48 hours and followed linearity between 0.5 -2.50 µg mL-1 of Pd (II) with an estimated molar absorptivity of 0.4 × 103 L mol-1 cm-1. The method's applicability was tested to determine Pd (II) from actual samples and complexes.

Scheme 7. 2-Hydroxy-1-naphthalene carboxaldehyde hydrazine carboxamide
In a potassium hydrogen phthalate-hydrochloric acid buffer of pH 4, Pd (II) was observed to form 1:2 yellow complex with 2-mercaptoethanol [47]. With a linear range of detection as 1.39-8.36 µg Pd (II) mL-1 and computed value of molar absorptivity and relative standard deviation as 2.2634 × 104 L mol-1 cm-1 and 0.42 %, respectively, the complex's maximum intensity was observed at 315 nm. The method's validity was tested by successfully determining Pd in Pd-BaCO3 and Pd-Charcoal catalysts.
Over the pH range of 4.8-6.5, 4-(2'-Furalideneimino)-3-methyl-5-mercapto-1,2,4-triazole [48] (Scheme 8) produced a yellow 1:1 complex of Pd (II) that was extractable into n-butanol. The complex's maximum absorbance was measured at 410 nm. The compound has a linearity obedience range 5-50 µg Pd (II) mL-1 and a molar absorption coefficient of 1.4 × 103 L mole-1 cm-1, respectively. RSD of the method was ± 0.55-1.0%. The method's applicability was tested by determining Pd (II) in synthetic mixes corresponding to alloys and catalysts.
 Scheme 8. 4-(2'-Furalideneimino)-3-methyl-5-mercapto-1,2,4-triazole
Scheme 8. 4-(2'-Furalideneimino)-3-methyl-5-mercapto-1,2,4-triazole
At room temperature and in 0.6-2.0 M hydrochloric acid medium, Pd (II) formed a stable ( > 24 h) yellow 1:1 complex with 4-(4'-fluorobenzylideneimino)-3-methyl-5-mercapto-1,2,4-triazole [49] (Scheme 9) that was soluble and extractable in chloroform exhibiting maximum colour intensity at 390 nm. B.L.range, ε and RSD of procedure were 4.13-17.5 µg Pd (II) mL-1, 5.404 × 103 L mol-1 cm-1 and 0.621%, respectively.
In the presence of sodium dodecylbenzene sulfonate and KH2PO4 -K2HPO4 buffer of pH 5.9-7.5, a colored reaction of 2-(p-carboxylphenylazo) benzothiazole [50] was studied with Pd in a divalent state to produce a 1:1 stable complex. The coloured complex was extracted with C8 cartridge, followed linearity in range 0.1-1.0 µg mL-1, and had a molar extinction coefficient of 1.61 × 105 L mol-1 cm-1 at 510 nm.

Scheme 9. 4-(4'-Fluorobenzylideneimino)-3-methyl-5-mercapto-1,2,4-triazole
A new analytical reagent, p-Methoxyphenylethane-1,2-dion-1-oxime [51], was employed to analyze Pd (II) spectrophotometrically where it formed a yellow 1:2 complex in the pH range 1.1-3.2, extractable into chloroform and showing maximum color intensity at 420 nm. B.L.range, ε and RSD (n=10), respectively were 1-20 µg Pd (II) mL-1, 3.087 × 104 L mol-1 cm-1 and 1.665%.
An aqueous solution of pH 0.0-4.0 and 0.1-1 M of acetic acid were used to extract a 1:2 combination of Pd (II) with 2-hydroxy-3-nitro-5-methylacetophenone oxime [52] quantitatively into chloroform. At 430 nm, the isolated complex's Beer's law range and molar absorption coefficient were 0.1-10 µg Pd (II) mL-1 and 1.5796 × 104 L mol-1 cm-1, respectively.
α-Benzildioxime [53] in thiocyanate was employed as a new complexing agent for spectrophotometric measurement of Pd (II), forming a yellow 1:2 complex that was extractable in chloroform and had a maximum colour intensity at 364 nm. The produced compound had a Beer's law range of 0-16 µg Pd (II) mL-1 and a molar absorptivity of 3.088 ×103 L mol-1 cm-1, respectively.
In an CH3COOH-CH3COONa buffer solution maintained at pH 5.5, a colour reaction resulted between Pd (II) and 2-(5-carboxy-1,3,4-triazolylazo)-5-diethylamino aniline [54] to give a stable 1:2 purple-red complex which was absorbed maximally at 585 nm. The compound had a Beer's law range of 0-1.6 mg Pd (II) L-1 and molar absorptivity of 1.2 × 104 L mol-1 cm-1, respectively.
At pH 6.0, an extractable yellow complex of Pd (II) was reported with 2-acetyl thiopyran thiocynate (ATPT) [55] (Scheme 10). The compound was extracted into n-hexane and measured at 520 nm for maximum absorption. The linearity range as obtained by the calibration graph was 5-40 µg Pd (II) mL-1 with the molar absorptivity of 0.2367 × 104 L.mol-1.cm-1.

Scheme 10. 2-Acetyl thiopyran thiocyanate (ATPT)
A blue complex of Pd (II) was obtained after its chromogenic reaction with bromo-sulfonazo-III [56] in a buffer medium of pH 2.9. λmax , Beer's law range and ε of the complex were 623.7 nm, 0-1.28 µg Pd (II) mL-1 and 6.375 ×104 L mol-1cm-1, respectively.
Trace level extraction and spectrophotometric detection of Pd (II) was reported with p-[4-(3,5-dimethylisoxazolyl)azophenylazo]calix(4)arene [57] where a sensitive and selective complex was formed that was extractable into 1,2-dichloroethane (30%), attained absorption maximum at 340 nm, had the linear range of 0.5-9.5 µg Pd (II) mL-1 , the calculated value of molar absorptivity and RSD of 1.73 × 104 L mol-1 cm-1and 0.68%, respectively.
A precise, fast, sensitive and selective method for micro-level detection of Pd (II) was proposed using hexamethyleneiminecarbodithioate [58] as a chelating agent. The 1:2 yellow complex was extracted into toluene from the acidic medium of pH 0.5-2.0, absorbed maximum at 435 nm and was stable for 7 days. Beer's law range and molar absorptivity were 0.2 to 0.8 µg mL-1 of Pd (II) and 0.754 × 104 L mol-1cm-1, respectively. The approach was utilized to accurately determine palladium in synthetic and actual materials.
Trace concentration of Pd (II) was determined spectrophotometrically as its stable extractable pink 1:2 (M:L) hexane -2,5-dione-bis-(ethylenediamine) [59] complex at pH 3.5. Extracted in chloroform, the compound achieved maximum colour intensity at 474 nm. The linearity range of the complex was 2-20 mg L-1 and molar absorptivity was 0.750 × 104 L.mol-1.cm-1.
Micro amounts of Pd (II) in Pt-Pd refined ores and artificial samples were determined with good precision (RSD ≤ 2.4%) by forming its extractable complex with polyethylene glycol-ammonium sulfate-DBOK-CPA [60], having a linear determination range being 0.09-2.0 mg L-1.
Pd (II) was quantitatively determined at pH 6.0 - 6.5 from a phosphate buffer solution as its 1:1 purple complex with 2-hydrazinylpyridine [61] (Scheme 11). More than 24 h, stable compound had absorption maximum at 510 nm, linear calibration range 1.06-9.00 µg Pd (II) mL-1, calculated value molar absorptivity and RSD as 2.978 × 103 L mol-1 cm-1 and 0.04-0.41%, respectively.

Scheme 11. 2-Hydrazinylpyridine
Using 5-chloro-8-hydroxy-7-iodoquinoline [62] as a chelating agent, a simple, rapid, sensitive, and selective technique for trace measurement of Pd (II) were presented. From 1 M H2SO4, the coloured compound was extracted in chloroform and absorbed at 450 nm. The range of Beer's law and molar absorptivities were 0-2.6 mg mL-1 and 0.9 × 104 L mol-1cm-1, respectively. The approach was used to determine the concentration of Pd in synthetic and actual samples.
As its extractable complex with 3-hydroxy-2-(2′-thienyl)-4H-chromen-4-one [63], Pd (II) was analyzed spectrophotometrically from the alkaline medium of pH 8.5-9.2. The yellow-coloured compound was 100% extracted into chloroform and attained maximum color intensity at 455 nm with Beer's law range, molar absorptivity and RSD as 0.01-0.1 µg Pd (II) mL-1 and 3.301 × 103 L.mol-1.cm-1, respectively.
Palladium (II) complexed with 3-hydroxy-2-[2'-(5'-methylthienyl)]-4-oxo-4H-1-benzopyran (HMTB) [64] (Scheme 12) in NaHCO3 supplied basic medium. Up to 1.3 µg Pd(II) mL-1, the alignment bends demonstrate linearity. The complex has a molar annihilation coefficient of 6.597×104 L mol-1 cm-1 at 430 nm and a separate detection limit of 0.042 μg.mL-1. Aside from the most favorable piece of leeway of swift and simple research by this optical density approach, the established method has been acceptably related for the assurance of the metal in various alloy samples, engineered blends, and industrial products.

Scheme 12. 3-Hydroxy-2-[2'-(5'-methylthienyl)]-4-oxo-4H-1-benzopyran (HMTB)
A simple, rapid, sensitive, and selective extractive spectrophotometric technique for the trace detection of palladium(II) from NaHCO3 medium was devised using another benzopyran derivative, 6-chloro-3-hydroxy-7-methyl-2-(2′-thienyl)-4-oxo-4H-1-benzopyran (CHMTB) [65] (Scheme 13) as a colouring agent. The metal complex was extracted quantitatively into ethyl acetate and absorbed at a maximum 415-426 nm wavelength. Over the Pd (II) concentration range of 0-2.6 µg mL-1, the 1:1 yellow complex obeyed Beer's law, with a correlation coefficient of 0.9998. When using spectrophotometric determination at 420 nm, the molar annihilation coefficient and Sandell's sensitivity were 6.173 × 104 L mol-1 cm-1 and 0.0017 µg Pd(II) cm-2, respectively. Os(VIII), Cr(VI), Mo(VI), V(V), Nb(V), Ce(IV), Se(IV), Zr(IV), Pt(IV), Ru(III), Ir(III), and Fe(III) do not conflict with the suggested approach. The approach was successfully used to determine Pd (II) in various substances, including water and palladium charcoal catalyst.

Scheme 13. 6-Chloro-3-hydroxy-7-methyl-2-(2′-thienyl)-4-oxo-4H-1-benzopyran
2.2 Solid phase extraction technique
Solid phase extraction (Figure 2) is applied as an extractive technique for separating the compounds, dissolved or suspended in a liquid mixture, based on their general properties. The technique is practically used in analytical laboratories to isolate analytes of interest from various samples, including urine, water, beverages, soil and animal tissue. As mentioned in the succeeding data, the approach is applied to the spectrophotometric determination of palladium.
Coupled with solid phase extraction (CPE) of the complex with a reversed-phase polymer-based C18 cartridge, 1-(2-Benzothiazolylazo)-2-hydroxy-3-naphthoic acid (BTAHN) [66] was applied as one of the most efficient reagents for spectrophotometric detection of the metal ion. The 1:2 deep violet M:L is formed in 5 M HCl medium and cetyltrimethyl ammonium bromide as surfactant. After extraction into isopentyl alcohol, 669 nm was the wavelength of maximum absorption. The complex obeyed linearity within 0.02-0.85 µg ml-1 palladium concentration range with the calculated molar absorptivity of 2.61 × 105 L mol-1 cm-1. RSD of the method was 1.06%. Low amounts of palladium in environmental samples were successfully determined using the method.
In the presence of the emulsifier OP, Pd (II) interacted with p-antipyrinylazo benzoic acid (AABA) [67] to create a 1:2 stable complex in an acetic acid-sodium acetate buffer with pH 3.5-5.5. The coloured complex was spectrophotometrically measured at 452 nm following extraction by C18 cartridge, fulfilled Beer's law in the range of 0.1-1.5 µg mL-1 Pd (II) and had a molar absorptivity of 1.22 × 105 L.mol-1 cm-1. Palladium in Pt/Pd catalysts was determined using this method.

Figure 2. Solid phase extraction technique
Solid phase extraction of the coloured complex between Pd in divalent state and 2-(2-quinolinylazo)-5-dimethylaminobenzoic acid [68] was studied with C18 cartridge. 1:2 Stable complex was formed with the reagent in acetic acid-sodium acetate buffer solution of pH 3.5 and in the presence of the surfactant CTMAB. Beer's law range was 0.01-1.5 mg Pd(II) L-1 at 630 nm.
A new determination method for Pd (II) was successfully studied in a medium of OP-SDBS and C3H5(COO)3HNa2 - NaOH buffer solution maintained at pH 5.0-6.3 using 1-(2'-benzothiazole)-3-(4'-carboxylbenzene) triazine (BTCBT) [69] as a reagent and extracting the thus formed colored complex by solid phase extraction with C8 cartridge. The 1:2 (M:L) complex exhibited maximum color intensity at 490 nm with Beer's law obedience range and molar absorptivity of 0.1-1.2 µg mL-1 of Pd (II) and 1.16 × 105 L mol-1 cm-1, respectively. To determine trace palladium in C-Pd catalysts, the method successfully was used.
In the presence of CTMAB and a pH 2.0 buffer solution, a very sensitive (ε = 7.79 × 104 L mol-1 cm-1) 1:2 complex of Pd (II) was generated with p-sulfobenzylidene - rhodanine (SBDR) [70] which was extracted with C18 cartridge and attained λmax at 535 nm. In the Pd concentration range of 0.01-2.0 µg mL-1, Beer's law held true. Palladium in cyanide residue was determined using this approach. Similar color reaction of Pd (II) was studied with p-rhodanineazobenzoic acid to produce a 1:1 stable complex having λmax = 500 nm, B.L.range = 0.1-1.0 0 µg mL-1 Pd (II) and ε = 1.36 × 105 L mol-1 cm-1).
In the presence of 0.05-0.5 M HCl and cetyltrimethyl ammonium bromide, 2-(2-Quinolylazo)-5-dimethylaminoaniline [71] (Scheme 14) was successfully employed as a chromogenic reagent for solid phase extraction and spectrophotometric detection of Pd (II). The reagent formed a 2:1 violet ring complex with a maximal color development time of 8 minutes after interacting with Pd (II). Solid phase extraction with a C18 cartridge enriched the produced compound, which remained stable for 10 hours. The complex attained maximum color intensity at 618 nm, obeyed linearity between 0.01 to 1.2 µg Pd(II) mL-1, had calculated values of ε and RSD as 1.41 × 105 L mol-1 cm-1and 2.05%, respectively. The same complex could be extracted in MCI-GEL resin [72] from a 0.2-2.0 M HCl medium following Beer's law at 0.01-1.5 mg Pd (II) L-1. Under similar conditions, a 1:3 purple complex of Pd (II) was formed with 2-(3,5-dichloropyridylazo)-5-dimethylaminoaniline [73] with λmax = 620 nm, B.L. range = 0.01-3.0 µg mL-1 of Pd (II) and RSD = 3.2%).

Scheme 14. 2-(2-Quinolylazo)-5-dimethylaminoaniline
2.3 Aqueous phase complexation technique
The technique (Figure 3) has been applied to satisfactory determination of the metal in form of various complexes as:
A stable 1:2 complex of Pd (II) was formed with 2-(2-quinolylazo)-1,5-diaminobenzene (QADAB) [74] in 0.2-3.0 M HClO4 in cetyltrimethyl ammonium bromide. The compound was based on the color reaction, and the solid phase extraction on a small column packed MCI-GEL reverse phase fine separation filler.
In the presence of cetyltrimethyl ammonium bromide, Pd (II) produced a violet-colored 1:2 complex with 2-(2-quinolylazo)-5-diethylaminobenzoic acid [75] in 0.05-0.5 M HCl solution (CTAB). When the compound was extracted into isopentyl alcohol, its absorption peak was measured at 628 nm. Beer's law held true in the range 0.01-1.2 µg Pd (II) mL-1 with molar extinction coefficient of the chelating complex as 1.43 × 105 L mol-1 cm-1. RSD for eleven replicates at the 0.2 µg L-1 level was 2.18%.

Figure 3. Aqueous phase complexation technique
2-(2-Quinolylazo)-5-diethylaminobenzoic acid [76] (Scheme 15) formed a 2:1 colored complex with Pd (II) in 0.5-2.5 M HCl and CTMAB medium. λmax was at 625 nm, the linear range was 0.01-0.6 μg Pd (II) mL-1 with molar absorption coefficient of 1.51 × 105 L mol−1 cm−1. RSD of the method was 0.75%. The successful detection ofsuccessfully detecting palladium established the method's validity in river water samples and catalysts.

Scheme 15. 2-(2-Quinolylazo)-5-diethylaminobenzoic acid
In aqueous medium, 4-(N′-(4-imino-2-oxo-thiazolidine-5-ylidene)-hydrazino)-benzoic acid [77] forms a compound with a maximum absorbance of 450 nm. 4.30 × 103 L·mol−1·cm−1 is the molar absorptivity. The linearity range of the developed technique is 0.64–10.64 µg Pd(II) mL−1. 0.23 µg mL−1 is the detection limit. Complex is free from the interference of Co(II), Ni(II), Zn(II), Fe(III), Cu(II), Al(III) and many anions. The proposed method has been effectively used to determine palladium in catalysts after being tested on model solutions. The results suggest that this method may be used to determine serial amounts of palladium in various objects.
In a miceller medium of neutral surfactant Triton X-100, Pd (II) formed bright yellow water-soluble 1:1 complex with 3,5-dimethoxy-4-hydroxy benzaldehydeisonicotinoylhydrazone [78] (Scheme 16) in an acidic buffer in the pH range 4.5-7.0. Stability of the complex was increased from 15 min to more than 6 h in the presence of Triton X-100. λmax, Beer's law range and molar absorptivity were 382 nm, 0.1064-2.1284 µg Pd (II) mL-1 and 2.44 × 104 L mol-1 cm-1, respectively. RSD of the method was 0.02 %. Cu (II) interfered seriously with the determination. In acidic buffer (pH 1.0-3.0; CH3COONa and concentrated HCl) Pd (II) reacted with pyridoxal thiosemicarbazone [79] (Scheme 17) to form a non-extractive 1:2 (M:L) pale yellow complex which was stable for 2 h at 420 nm. Beer's law range, molar absorptivity, and relative standard deviation of the method were 0.0-10.0 µg Pd (II) mL-1, 1.63 × 104 L mol-1 cm-1 and 2.47%, respectively.
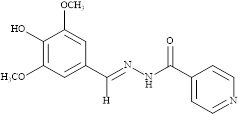
Scheme 16. 3, 5-Dimethoxy-4-hydroxy benzaldehydeisonicotinoylhydrazone(DMHBIH)

Scheme 17. Pyridoxal thiosemicarbazone
In an aqueous medium at pH 10, Pd (II) interacted with thioglycolic acid [80] to generate a yellow 1:2 complex. The compound remained stable for 24 h and absorbed light most efficiently at 384 nm. With a molar extinction coefficient of 2.0692 × 104 L mol-1 cm-1, the compound demonstrated linearity to 8 mg L-1 of Pd (II)
Using 2,5-dimercapto-1,3,4 thiadiazole [81] in the presence of the surfactant Triton X-100, a simple, sensitive and selective spectrophotometric technique for the trace detection of Pd (II) in alloys and minerals was presented. Linearity of the 1:1 colored complex was found in the Pd (II) concentration range 0.20-2.5 µg mL-1, with a molar absorption value of 3.03 × 104 L mol-1 cm-1 at 375 nm.
With a molar absorptivity of 4.46 × 105 L mol-1 cm-1, 2,4-Dichlorophenylfluorone [82] produced a stable combination with Pd (II) in mild acid. The approach was effectively used to determine the metal content of Pd-minerals and Pd-catalysts.
At pH 4, a 1:2 yellow-colored complex between Pd (II) and L-cystine [83] was produced which was investigated spectrophotometrically demonstrating maximum absorbance at 369 nm and obeying Beer's law over the concentration range 2.12-16.9 μg L-1 of palladium (II). The yellow-colored complex's molar absorption coefficient and sensitivity were 2.69 ×104 L mol-1 cm-1 and 7.89 ×10-4 μg cm-2, respectively. Pd (II) in synthetic alloy samples and hydrogenation catalyst samples were successfully analyzed using this approach.
The complex formation was seen as a reaction between Pd (II) and cefixime at room temperature and in the presence of a Na2HPO4-citric acid buffer solution [84] with a pH of 2.6 in a methanol-distilled water medium. The complex's maximum absorption wavelength was 352 nm with the linear range 0.7502–16.5004 µg mL-1. The produced compound has an apparent molar absorptivity of 1.224 × 104 L mol-1 cm-1. The method's applicability was demonstrated in Pd (II) measurement in synthetic mixtures and automotive workshop area samples.
The new analytical reagent, pyridoxal thiosemicarbazone (PTSC) [85] was employed for direct non-extractive spectrophotometric measurement of palladium (II) hence giving a highly elementary and selective method in an acidic medium of pH 2. The M:L∷1:2 pale yellow compound exhibited maximum intensity of colour at 420 nm remaining consistent for 2 h. [ε = 1.63 × 104 L mol-1 cm-1, S = 0.635 μg cm-2, B.L. range = 0.9- 10.0 μg Pd(II) ml-1]. The approach successfully analyzed palladium in a variety of hydrogenation catalysts.
- Conclusions
Several techniques, particularly gravimetric, titrimetric and the instrumental like AAS, X-ray fluorescence, NAA, ICP-emission spectroscopy and UV-VIS spectrophotometry can be used for trace determination of the analyte from matrix components. However, the UV-VIS spectrophotometric determination method finds wide applicability and makes this attractive approach to pursue. The technique offers the advantage of rapid determination, easy handling, low-cost equipment, superior sensitivity, selectivity and precision compared to the other similar techniques used for the micro analysis of the element thus ensuring a dominant technique in the field of instrumental methods of elemental analysis. The recent studies on the spectrophotometric methods of palladium determination are based upon increasing use of the element in various fields, including jewelry, electrical and dental appliances, alloys and particularly catalysis. The article updates the methods for trace determination of palladium in different synthetic and industrial samples and will be enormously advantageous to analysts.
)